
基于活性金电极上硫代水杨酸自组装单分子层的电化学表面增强拉曼光谱
2013-10-18 05:27刘文涵袁荣辉滕渊洁马淳安
物理化学学报 2013年12期
刘文涵 袁荣辉 滕渊洁 马淳安
(浙江工业大学化学工程与材料学院,绿色化学合成技术国家重点实验室培育基地,杭州 310032)
1 引言
表面增强拉曼散射(SERS)光谱凭借其极高的检测灵敏度,在表面结构、表面吸附以及界面特性等领域1-3得到了广泛的运用,已使其成为在分子水平上表征金属/溶液界面的电化学过程最为有效的技术.
分子自组装技术可在基底表面形成稳定有序、可控、均一整齐的自组装单分子层(SAMs),已广泛应用于制备功能性薄膜、分子识别、功能化修饰以及电极表面改性等研究中,特别是硫醇化合物的SAMs研究较为突出.4-6含巯基的分子在金表面成膜已被应用于传感器、修饰电极等研究领域,7,8当其吸附到金电极表面时,可以形成稳定的单分子层,使得修饰电极呈现出新的电化学特性.9,10硫醇类化合物在经过特殊处理的金表面会产生很强的特征拉曼光谱信号.11-13硫代水杨酸(TSA)为芳香族硫醇化合物,芳基硫醇分子具有高度的各向异性,且TSA较其他对位的巯基化合物制备容易、成本较低.以活性金作基底材料相比活性银不容易在空气中形成氧化膜,也不易被电腐蚀.因此,对于金表面的TSA自组装单层的研究具有一定的理论意义和潜在的应用前景.
本文采用电化学活化金电极表面的方法,制备具有表面活性的粗糙金电极,利用原位电化学表面增强拉曼光谱(EC-SERS)技术,在分子水平上表征电吸附在金表面的TSA单分子层,研究了苯环上带有的硫醇端基的振动光谱特性.探讨了不同电位等条件下对TSA溶液EC-SERS的影响.并通过计算TSA在不同pH条件下存在形式的分布以及施加高负电位前后的增强因子(EF),进而研究了TSA在金电极上的电化学吸附取向并对机理进行了探讨.
2 实验部分
2.1 仪器与试剂
LabRAM HR UV 800激光显微拉曼光谱仪(法国JOBIN YVON公司);激发光源:632.81 nm He-Ne激光器;共焦孔径300 μm,光栅刻线数600 lines·mm-1,物镜:50倍长焦距镜头.CHI660B型电化学工作站(上海辰华仪器有限公司).PHSJ-4A型实验室pH计(上海精密科学仪器有限公司).直径3 mm金盘电极(天津艾达恒晟科技发展有限公司).
硫代水杨酸(99%,Aladdin试剂有限公司),其它试剂均为分析纯及以上.粒径30-50 nm的氧化铝抛光粉,水为18.3 MΩ·cm超纯水,由HUMAN UP900型超纯水器(韩国HUMAN公司)制得.
2.2 金电极电化学活化
未“活化”的金表面不能产生SERS效应,需先进行粗糙活化处理:先将金圆盘电极在金相砂纸上打磨,再将细Al2O3抛光粉与水混合成悬浮液,在定性滤纸上将电极抛成镜面,依次用无水乙醇、超纯水超声各2 min,获得洁净的电极表面.以金盘电极为工作电极,铂丝为辅助电极,饱和甘汞电极(SCE)为参比电极,组成三电极体系.0.3 mol·L-1硫酸为支持电解质底液,以100 mV·s-1的扫描速率在-0.2 V至上限1.6 V的电位区间内反复进行电位扫描,以增加电极表面的活化位点,14待循环伏安特征稳定后,换用0.1 mol·L-1KCl作电解质底液,在电位区间-0.1-1.3 V范围内,以10 mV·s-1扫描速率循环扫描3周,再用超纯水清洗电极.金盘电极经电化学氧化还原(ORC)处理,获得一层暗色且具有SERS活性的表面.
2.3 激光拉曼光谱和SERS光谱测定
常规拉曼光谱(NRS)测定:将经无水乙醇溶解并重结晶后的固体样品置于载玻片上,于激光显微拉曼光谱仪载物台的物镜视野下,通过计算机的影像窗口调整焦距,使激光聚焦于样品表面,进行NRS测试.扫描范围200-2800 cm-1,光谱信号采集时间10 s,积分2次平均.
液相现场单分子层SERS测定:以活性金电极作为基底材料,于光谱电化学池内,在不施加电位的情况下,电极表面在1×10-3mol·L-1、pH=3.2的TSA待测溶液中浸置90 min进行自组装,并控制电极表面的液面厚度在1 mm内进行SERS光谱扫描.扫描范围200-2800 cm-1,采集时间50 s,积分2次平均.
浸饰膜SERS测定:配制不同pH值的1×10-3mol·L-1TSA溶液,将活性金电极浸入不同pH值的TSA溶液中,浸置90 min进行自组装,取出电极在高纯N2氛围中干燥,使TSA充分吸附附着在电极表面,进行修饰膜的SERS光谱检测,扫描范围200-1700 cm-1,采集时间10 s,积分2次平均,在获取SERS峰强时,需测定电极表面3个不同位置点的数值取平均,峰强度数据由软件Labspec读出.
2.4 现场EC-SERS测定
因酸性体系TSA得到的SERS效应强于碱性体系,TSA在pH<3时有形成悬浊液而未能很好测得.故选取以0.1 mol·L-1KCl作底液,TSA浓度为1×10-3mol·L-1、pH=3.2的溶液置于光谱电化学池内,控制其电极表面的液面厚度在1 mm内,连接三电极体系,使激光聚焦在电极表面,进行原位EC-SERS测试,测定不同通电富集作用时间以及不同电位下TSA分子的EC-SERS.扫描范围200-1700 cm-1,采集时间50 s,积分2次平均.
3 结果与讨论
3.1 TSA在活性Au表面的化学吸附
为考察TSA在活性Au上的吸附作用,测定了1×10-3mol·L-1TSA溶液、pH=3.2的SERS(图1(b))与TSA晶体的NRS(图1(a))对比.用激光拉曼光谱直接测定1×10-3mol·L-1TSA溶液,由于浓度太低而溶液的NRS信号很不明显,因而在1×10-3mol·L-1TSA溶液中的Au表面测得的拉曼光谱,可认为是由于TSA分子吸附键合在Au表面后产生的SERS光谱.非晶形粉末TSA经重结晶后的NRS以及TSA在活性Au电极上的SERS谱峰归属列于表1.
从图1可以看出,Au表面的SERS相比TSA晶体的NRS发生一些变化,TSA原有的S―H特征摇摆振动弱峰(311 cm-1)消失,替代并新产生了S―Au峰(269 cm-1),15,16同时归属S―H伸缩振动的强峰(2520 cm-1)亦消失.SERS出现了多个新峰,NRS中共有峰的拉曼位移在SERS中出现了普遍向低波数移动的现象(表1).表明TSA已在“粗糙”的Au表面发生了化学吸附并促进了拉曼的化学增强等作用.
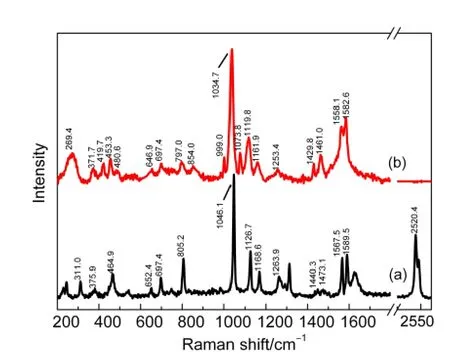
图1 TSA晶体NRS(a)与1×10-3 mol·L-1 TSA溶液在活性Au表面的SERS光谱(b)Fig.1 Normal Raman spectroscopy(NRS)of TSAcrystal sample(a)and SERS spectrum of 1×10-3 mol·L-1 TSA solution on activated Au surface(b)
3.2 不同pH值下的浸饰TSA单层膜的SERS光谱
将活性Au电极浸入不同pH值的TSA溶液中,静置90 min进行自组装,再取出电极在高纯氮中干燥,使TSA充分吸附附着在Au表面形成浸饰膜,进行SERS测试,结果见图2A.
当测试Au表面上不同pH的TSA时,均可观察到S―Au的伸缩振动峰,表明TSA通过S―H失去质子与Au形成强的S―Au键吸附于电极表面成“膜”,随着pH的上升,苯环的呼吸特征峰(1035 cm-1)强度逐渐变弱;其它基团的特征峰,如453 cm-1的双取代苯振动、481 cm-1的C―S面外弯曲振动、647 cm-1的1,2-双取代苯骨架振动以及697 cm-1归属C―S伸缩振动等峰高亦随之降低.
以最强峰苯的环呼吸特征峰强与pH作关系图(图2B).可以看出,随着pH增加,SERS的峰强信号逐渐减弱,其变化趋势以pH 5-6为分界,表现为不同的2个阶段,在pH 6及以上时峰强I2随着pH的变化的下降速率即斜率为pH 3-5时I1的2倍,表明SERS效应下降速率增加了1倍.当pH>11时谱峰强度已很弱,表明碱性体系已不利于TSA在Au界面上的成键结合或不利于增强效应的产生,表观结果是OH-的存在会加速阻止TSA的SERS效应.
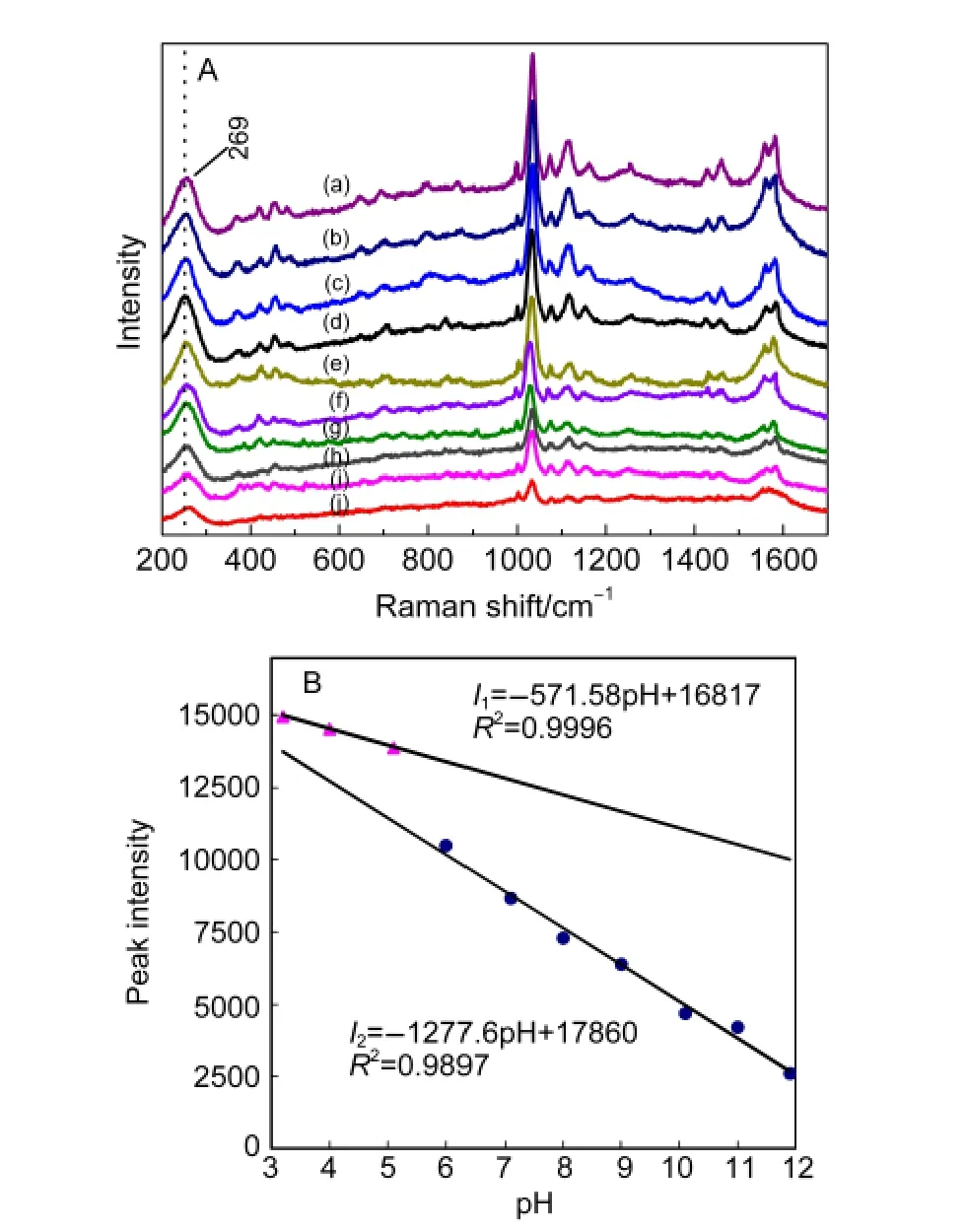
图2 不同pH值对TSA修饰膜的SERS光谱(A)及苯环呼吸特征峰强(B)的影响Fig.2 SERS spectra of gold electrodes modified with TSA(A)and intensity of 1035 cm-1 SERS peak at different pH values(B)
3.3 TSA在活性Au表面的EC-SERS
为了进一步考察Au电极表面的TSA在通电条件下的吸附性能,及TSA与Au基底在不同的恒电位条件下的相互作用,分别探讨了不同电富集时间以及不同电位条件对EC-SERS的影响.
由于电化学ORC得到的粗糙Au表面,在较高电位下仍然会发生电化学溶出现象,粗糙Au原子变为Au离子而失去SERS效应.因而需先考察其电极行为,以探讨合适的电化学吸附所需的电位.电极经抛光、超声清洗、活化处理,将Au工作电极浸入0.1 mol·L-1KCl溶液中,使用三电极体系对自组装TSA修饰膜后的Au电极进行阳极极化曲线测试,扫描速率为10 mV·s-1.按同一方法测定未经修饰的活性Au工作电极在0.1 mol·L-1KCl底液的阳极溶出电位作为本底对比,电化学实验均在室温下进行((23±2)°C).结果表明,电化学溶出腐蚀电位均在0.8 V以上.为保证ORC活化后的金基底的ECSERS活性和同时作为工作电极而不被电氧化,其所施加的正电位不应超过0.8 V.
3.3.1 不同电富集时间对EC-SERS的影响
将活性Au电极浸没于以0.1 mol·L-1KCl作支持电解质底液,TSA为1×10-3mol·L-1、pH=3.2的待测液中.测定了0.1 V电位下,在10-100 s(间隔10 s)的不同电预富集时间的EC-SERS,结果如图3A.并以苯的环呼吸特征峰强与电富集时间作关系图(图3B).
可看出,在一定条件下,通电吸附累积到70 s时,峰强最大,继续延长时间,EC-SERS效应反而下降.推测其可能的机理是随着电富集时间的延长,Au电极表面吸附的TSA分子或分子层逐渐增多,反而阻止了EC-SERS作用,表明在1×10-3mol·L-1数量级时,电富集70 s下TSA组装形成SAMs有最佳的效果.另外可能,S―H与Au表面形成S―Au键合后,C6H4(COOH)SH团簇向外层空间伸展,随着电累积时间的增加,分子内及分子间的彼此接近,产生“拥堵”而引起空间阻碍,形成分子的空间位阻效应,从而减弱了其吸收激发光能量的同时阻碍了振动所散射出的拉曼信号,因而EC-SERS信号减弱.

表1 TSA晶体的NRS,TSA在金电极上的SERS与EC-SERS位移(cm-1)对比及谱峰归属15,17,18Table 1 Comparison of Raman frequencies(cm-1),intensities and vibrational modes of TSAin NRS of crystal,SERS,and EC-SERS15,17,18
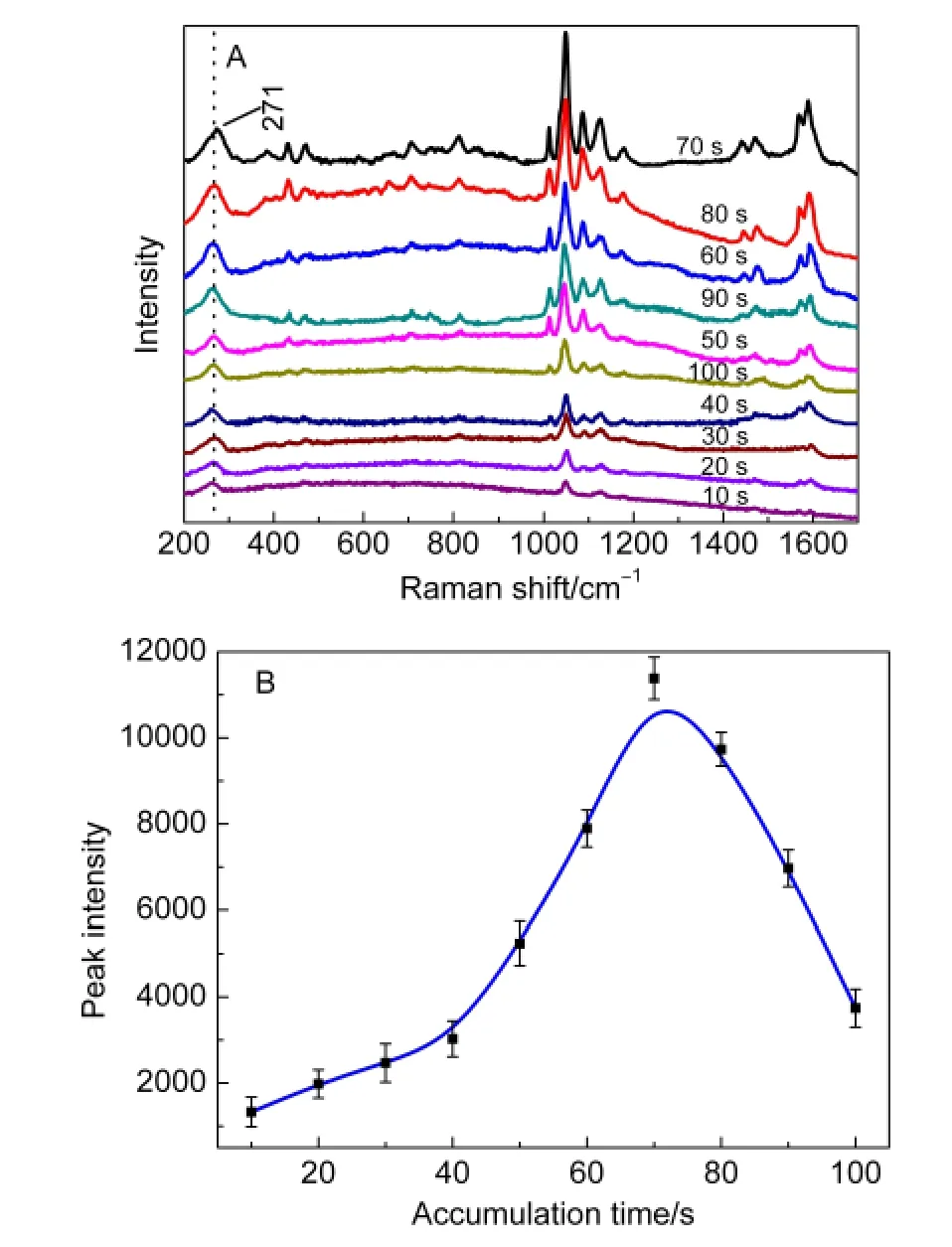
图3 0.1 V电位(vs SCE)下不同电富集时间对TSA的EC-SERS(A)及苯环呼吸特征峰强(B)的影响Fig.3 EC-SERS of TSA(A)and intensity of 1036 cm-1 peak with different accumulation time at 0.1 V(vs SCE)(B)
3.3.2 不同电位与EC-SERS的关系
测定了在不同的恒电位条件下,以0.1 mol·L-1KCl为底液,1×10-3mol·L-1、pH 3.2的TSA溶液,在电化学预富集70 s时间下的EC-SERS,见图4.TSA在Au电极上的SERS以及EC-SERS位移对比和具体谱峰归属列于表1.从表1的对比中可以看出,采用单纯溶液富集与电化学富集组装,S―Au的伸缩振动向高波数发生了移动(269→271 cm-1),同时增加了一些特征谱峰,共有的谱峰有所增强(w→m,m→s,s→vs),且基本都发生了右移,表明Au表面施加的电场,在活性Au吸附C6H4(COOH)SH时起到了一定的电化学增强的作用.
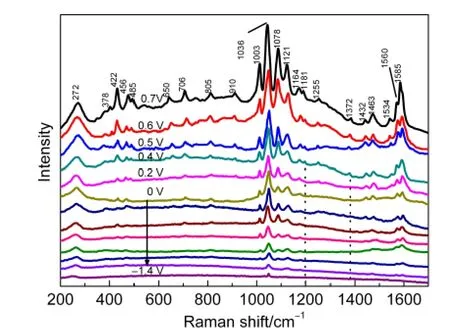
图4 不同电位条件下金电极表面TSA单分子层的EC-SERSFig.4 EC-SERS of TSAmonolayer on gold electrode at different potentials from+0.7 to-1.4 V(vs SCE)
同时考察了在电位0.2 V下,通电预富集70 s再断开电路后,测定10、200、300、500 s时的EC-SERS信号,结果表明谱峰信号及位移的变化不大,仅峰强略有增加.
3.4 TSA在不同pH值条件下的存在形式及机理探讨
TSA(C7H6O2S)在水中存在着离解平衡,其p Ka1值19为4.05,在水中不存在p Ka2使C7H5O2S-继续离解为C7H4O2S2-.图5是根据酸碱常数,计算与绘制TSA在不同pH值时的形态分布.δ1为中性分子C6H4(COOH)SH的分布分数,δ2为负一价离子C6H4(COO-)SH的分布分数(δ2=1-δ1).TSA为弱酸,当pH在3以下时,基本以中性分子形式存在,解释了TSA在pH<3时有形成悬浊液的现象.pH 4.05时中性分子与负一价离子的数目相等;随着pH升高,δ1含量逐渐减少,当pH升至6时,中性分子数目减少至1/100,此后基本以δ2为主.
3.4.1 TSA在Au表面的化学吸附机理
结合图2、5可以看出,溶液的酸碱度对TSA在Au表面的SERS光谱有一定的影响,随着pH值及TSA分子状态的改变,基本可分2个阶段.
溶液pH在5以下时,酸度较大,氢离子数目较多,羧基数目还较多,COOH中C=O、C―O诱导吸/拉电子能力较强(图6(a)),故S的电子云密度减小,对H的束缚力减弱,容易失去H与活性Au作用成键,与基底Au形成“强烈”吸附,自组装成单层膜而产生SERS且信号较强.
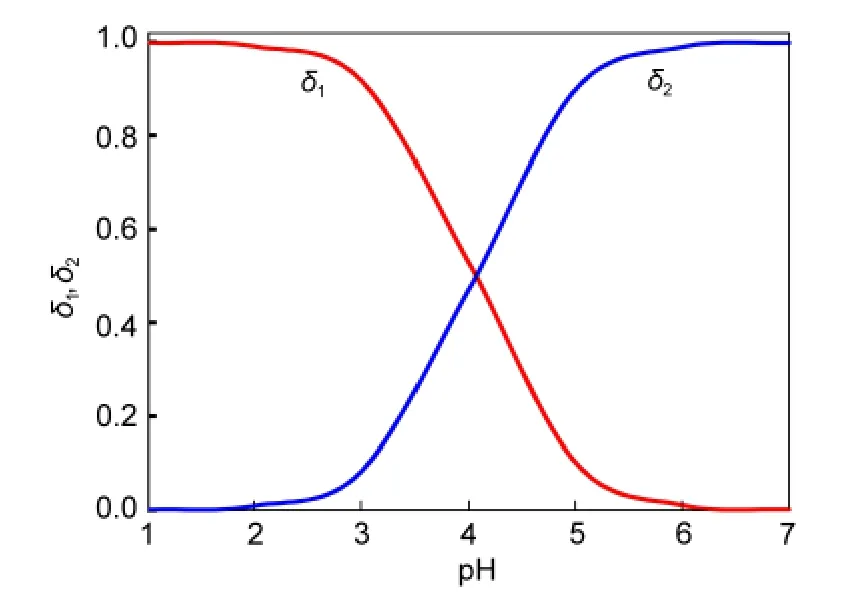
图5 TSA的分布分数(δ1,δ2)与溶液pH的关系Fig.5 Distribution fraction(δ1,δ2)of TSA at different pH values
当溶液逐渐趋向碱性时,TSA发生解离,在pH 6以上时,基本以负一价δ2存在,所产生的COO-有很强的供电子能力,苯环和羰基π-π共轭产生给电子作用,使S原子吸引H的能力增强(图6(b)),使得S―H中的H不易脱去,造成S―Au较难成键,因而信号减小.随着溶液中OH-的增加,SERS峰强发生了线性的下降,此时是否OH-也参与了竞争吸附,而排斥了TSA在Au表面的吸附,抑或是强碱性条件下,大量产生的COO-酸根与活性Au部分成键,使TSA的吸附方式发生了改变,垂直吸附(见图7(a))转化为“斜躺”式(图7(c)),而造成SERS效应减弱.
3.4.2 TSA在Au电极表面的电化学吸附机理及增强因子计算
在pH 3.2对1×10-3mol·L-1TSA溶液进行ECSERS测定时,其溶液中有δ1和δ2两种形态,且δ1的含量占87.6%,而δ2只占12.4%;故pH 3.2的TSA在Au上起到界面相互作用的主要形态是δ1.
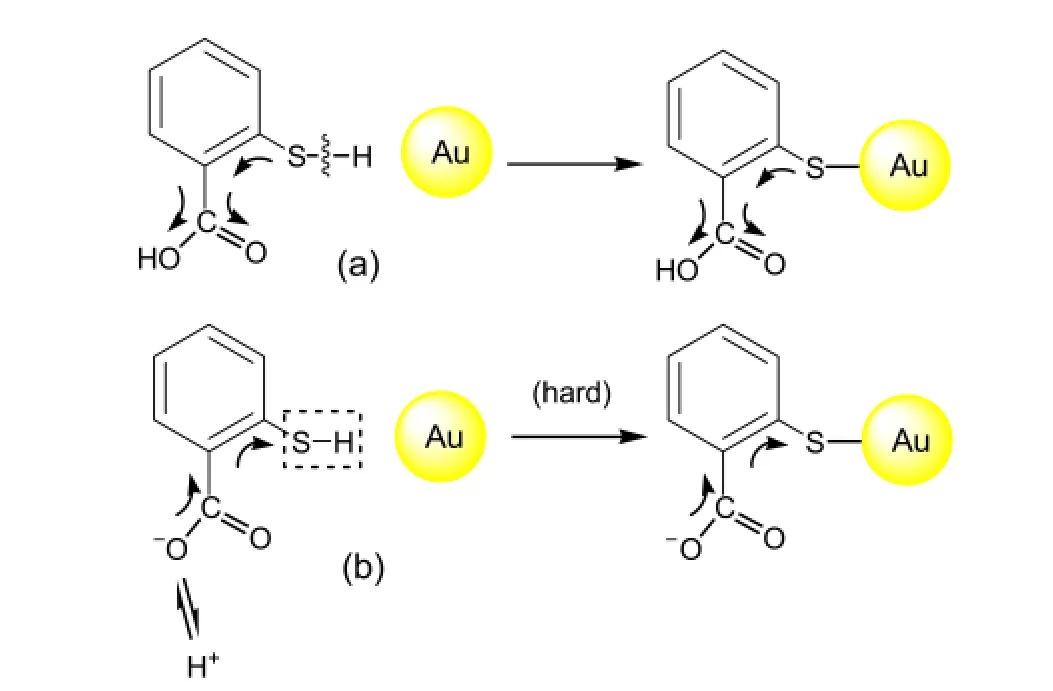
图6 TSA在不同酸碱下的化学吸附作用机理Fig.6 Chemical absorption mechanisms of TSA monolayer at acid(a)and base(b)conditions
从图4可以看出,峰强度随着电位正移而增大,0.7 V时EC-SERS信号最强.根据SERS表面选择定律,20,21直接作用于或靠近SERS基底的分子或基团,其在垂直方向上有最大分量的振动模式可得到最大增强,且越垂直于SERS基底的振动,增强效应越明显.因此在所加电位不产生电化学溶出基底材料的前提下,S原子已与基底Au发生了一定的键合作用,且在外加电位越正时,振动增强作用越强,越有效;或亦表明电位越正,在电场的作用下垂直方向排列的分子比例越多,因而归属S―Au伸缩振动的271 cm-1峰强和相应的其它EC-SERS峰越能明显地呈现出来.
EC-SERS与SERS相互对比,可以看出施加电位≥0.4 V时,EC-SERS多出了归属苯C―H面内变形的谱峰(1180和1372 cm-1)(表1),此外归属C=C伸缩振动的谱峰(1534 cm-1)也被检测到;同时苯C―H面内弯曲(1121,1164 cm-1)峰的强度显著增大(图4);而分子面内和面外振动的峰强度比值越大,即面内振动峰相对较强时,也说明了分子越垂直于基底.另外EC-SERS中归属S―H伸缩振动峰2520 cm-1没有出现(表1),同样认为溶液本底中的δ1通过硫醇端基失去质子,与Au形成S―Au吸附键合产生了EC-SERS,其垂直吸附作用于带正电的Au电极表面(图7(a)),而羧基向外暴露.而1255 cm-1归属C―O伸缩振动的谱峰很弱,可推断COOH与Au基底有一些距离.
此外,苯环上带负电荷的COO-也可作为另一个作用位点接近带正电的Au电极表面形成“静电吸附”(图7(b),图7(c)).从而认为少量的δ2是通过巯基和羧酸根COO-以较斜躺的方式自组装在电化学活化的带正电的Au表面.

图7 金电极表面TSA在正电位作用下的吸附取向Fig.7 Proposed orientations of TSAadsorbed on gold electrode at positive potential
另一方面,由于在负电位时,带负电的Au会与COO-相互排斥,而溶液中δ2约为12.4%,所产生的信号基本可以忽略,只需考虑占大多数的δ1形态在带负电位的电极表面的相互作用.从图4中EC-SERS信号强度是随负电位大小的变化而改变,可知吸附取向也与负电位值有直接的影响关系.当施加电位负移时,最强峰苯环呼吸振动的强度逐渐减弱,推测是通过S原子吸附在Au表面的TSA分子中的苯环由垂直方式逐渐趋于倾斜.同时与苯环相关的谱峰,如805 cm-1归属取代苯C―H面外弯曲、C―C伸缩振动(1432,1463 cm-1)和苯衍生物的环伸缩双峰(1560,1585 cm-1)等峰强都逐渐减弱,表明苯环有逐渐脱离基底表面的趋势.由于在此浓度下,常规拉曼光谱检测不到拉曼信号,所产生的信号均由SERS产生,因而TSA分子的拉曼光谱均会随分子吸附的改变而发生变化.当外加电位至-1.0 V时,信号继续变弱,且峰强已很不明显了,推测δ1在带负电的Au上开始部分脱离或发生吸附取向的变化.从伏安扫描图中表明,S―Au的还原峰起始电位出现在-0.91 V,峰值在-1.07 V左右.当电位进一步负移至-1.4 V时,EC-SERS谱峰基本上消失,故可推断活性Au电极表面的TSA,在高负电位下发生了还原脱附.22,18,23其吸附取向随负电位增大的变化过程如图8所示.
同时,经电化学粗糙化处理后的电极,其表面具有亚稳态纳米结构,基于EF与电位的密切相关性,24不同电位对EF的影响同样值得关注.
本文选用垂直吸附取向的TSA(pH=3.2)作为探针分子来进行活性Au表面的EF估算,EF的求取方法25-27见式(1):

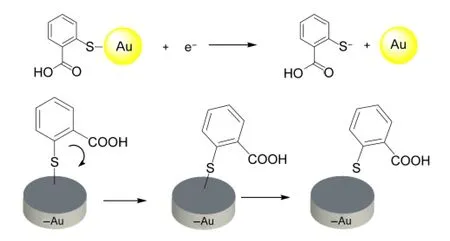
图8 金电极表面TSA的吸附取向随负电位增大的变化过程Fig.8 Orientation change of TSAmonolayer covered on gold electrode with the increase of negative potential
其中,Isurf为吸附在基底上TSA分子的SERS特征峰强,Nsurf表示活性Au基底上激光所照射到的TSA分子数,Ibulk为溶液中TSA的NRS特征峰强,Nbulk表示溶液中NRS测试时激光所照射的有效TSA探针分子数.此处,Isurf和Ibulk可分别从1×10-3mol·L-1TSA的SERS及0.5 mol·L-1TSA本体溶液的NRS谱图中,对应选择苯环的呼吸特征峰强直接得到(Ibulk=751.63).而Nsurf和Nbulk需通过计算,结合3.3.1节,判断浸置90 min、电预富集70 s进行自组装的1×10-3mol·L-1TSA分子为饱和吸附,则Nsurf可由式(2)表达:

σ为单个吸附分子占的表面积,金基底上的σ值为常数28(σAu=0.27 nm2).A为激光聚焦焦点的有效面积.R为活性Au基底的粗糙因子,可采用Trasatti和Petrii29的方法计算,通过比较Au在0.3 mol·L-1硫酸底液中,ORC活化前表面平整的基底上Au3+还原峰电量Q(对应Au3+还原曲线的积分面积)以及活化后粗糙表面的Au3+还原峰电量(Q*)可得,如(3)式:

A*为ORC活化后工作电极的有效面积,Ar为直径3 mm金盘电极的几何面积,求得R=1.104.
式(1)中的Nbulk需在考虑共焦显微拉曼的共焦特征的基础上进行计算,30见式(4):

C*为TSA本体溶液的浓度(C*=0.5 mol·L-1),NA为阿伏伽德罗常数(NA=6.02×1023mol-1).h为激光束照射到溶液中理想聚焦面的有效深度,可按Álvarez-Puebla等31的方法来计算,如式(5)所示:

n为环境媒介反射因子,即波长λ=632.81 nm的激光从空气介质到溶液中的折射率(n=4/3),N.A.为50倍长焦距物镜的数值孔径(N.A.=0.55),通过计算得到本文所用激光显微拉曼光谱仪的h=2.789 μm.
因0.5 mol·L-1TSA本体溶液NRS的光谱信号采集时间为80 s,在对本体溶液不同的采集时间进行折算校正的基础上,综合式(1)、(2)、(4)进一步推导可得:

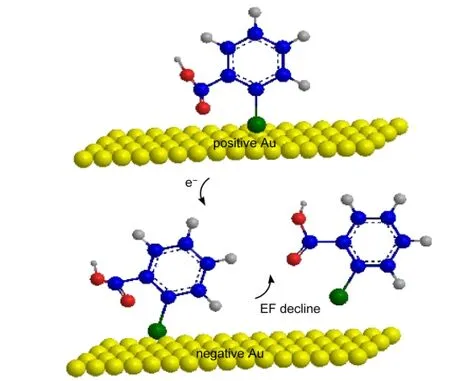
图9 电位条件从正到负过程中活性金电极上TSA单分子层的吸附与还原脱附过程Fig.9 Adsorption and reductive desorption process of TSAmonolayer on activated gold electrode potential varying from positive to negative
则1×10-3mol·L-1TSA在采集时间50 s的条件下,由式(6)最后求得施加0 V时的EF0=4.74×103,0.4 V下的EF0.4=8.26×103,-1.4 V下的EF-1.4=319,施加电位-1.8 V之后再回至0.4 V的EF-1.8/0.4=137,说明加了高负电位后再加正电位,其EF仍然较小,表明电极的拉曼增强活性已降低.
结果表明,SERS基底在高负电位的作用下,EF明显减小,TSA发生了还原脱附,此时,因电位再回复至正电位,EF值仍较小,表现出电极性能已发生了改变,并不可逆地降低或失去了拉曼光谱增强的活性.TSA单分子层在电位条件从正到负过程中的吸附与还原脱附过程见图9.
4 结论
在经电化学ORC粗糙活化处理后的Au电极表面,TSA通过断裂S―H键与Au形成S―Au键而吸附,在活性Au表面进行自组装形成SAMs,并产生SERS以及EC-SERS.在酸性介质中,由于COOH中C=O、C―O诱导吸/拉电子的作用,比在碱性条件下更易自组装成单层膜,而产生较强的SERS效应.在现场EC-SERS中,加0.7 V和电预富集70 s下的信号最强,随着电位负移,尤其在还原电位后,EF不可逆的减小,信号逐渐减弱,直至基本消失.表明TSA在Au表面的自组装过程的SERS信号受到电位和吸附时间的影响,TSA分子在Au表面排布状态会随施加条件的改变而发生变化.在-1.4 V时发生了不可逆的还原/解吸,同时亦造成Au表面活性的降低,使EC-SERS信号消失.另外作者还推测,是否单分子层的排布、电磁场的物理抑制、电荷转移机制等其它原因,也造成了一定的影响?这些都有待进一步计算和探讨.
(1)Titus,E.J.;Weber,M.L.;Stranahan,S.M.;Willets,K.A.Nano Lett.2012,12(10),5103.doi:10.1021/nl3017779
(2)Su,Q.Q.;Ma,X.Y.;Dong,J.;Jiang,C.Y.;Qian,W.P.ACS Appl.Mater.Interfaces 2011,3(6),1873.doi:10.1021/am200057f
(3)Freye,C.E.;Crane,N.A.;Kirchner,T.B.;Sepaniak,M.J.Anal.Chem.2013,85(8),3991.doi:10.1021/ac303710q
(4)Wang,Y.Q.;Yan,B.;Chen,L.X.Chem.Rev.2013,113(3),1391.doi:10.1021/cr300120g
(5)Chu,H.;Yang,H.F.;Huan,S.Y.;Shen,G.L.;Yu,R.Q.J.Phys.Chem.B 2006,110(11),5490.doi:10.1021/jp053914m
(6)Kho,K.W.;Dinish,U.S.;Kumar,A.;Olivo,M.ACS Nano 2012,6(6),4892.doi:10.1021/nn300352b
(7)Monnell,J.D.;Stapleton,J.J.;Dirk,S.M.;Reinerth,W.A.;Tour,J.M.;Allara,D.L.;Weiss,P.S.J.Phys.Chem.B 2005,109(43),20343.doi:10.1021/jp044186q
(8)Sumner,J.J.;Creager,S.E.J.Phys.Chem.B 2001,105(37),8739.doi:10.1021/jp011229j
(9)Napper,A.M.;Liu,H.Y.;Waldeck,D.H.J.Phys.Chem.B 2001,105(32),7699.doi:10.1021/jp0105140
(10)Bertin,P.A.;Ahrens,M.J.;Bhavsar,K.;Georganopoulou,D.;Wunder,M.;Blackburn,G.F.;Meade,T.Org.Lett.2010,12(15),3372.doi:10.1021/ol101180r
(11)Ameer,F.S.;Hu,W.F.;Ansar,S.M.;Siriwardana,K.;Collier,W.E.;Zou,S.L.;Zhang,D.M.J.Phys.Chem.C 2013,117(7),3484.
(12)Dasary,S.S.R.;Singh,A.K.;Senapati,D.;Yu,H.T.;Ray,P.C.J.Am.Chem.Soc.2009,131(38),13806.doi:10.1021/ja905134d
(13)Xie,W.;Walkenfort,B.;Schlu cker,S.J.Am.Chem.Soc.2013,135(5),1657.doi:10.1021/ja309074a
(14)Yang,Y.C.;Xia,Y.;Huang,W.;Zheng,J.F.;Li,Z.L.J.Solid State Electrochem.2012,16(4),1733.doi:10.1007/s10008-011-1600-8
(15)Ohta,N.;Yagi,I.J.Phys.Chem.C 2008,112(45),17603.doi:10.1021/jp806599r
(16)Carron,K.T.;Hurley,L.G.J.Phys.Chem.1991,95(24),9979.doi:10.1021/j100177a068
(17)Ke,Y.K.;Dong,H.R.Handbook of Analytical Chemistry:Spectrial Analysis;Chemical Industry Press:Beijing,1998;pp 1153-1160.[柯以侃,董慧茹.分析化学手册:光谱分析.北京:化学工业出版社,1998:1153-1160.]
(18)Schalnat,M.C.;Pemberton,J.E.Langmuir 2010,26(14),11862.doi:10.1021/la1010314
(19)Dean,J.A.Langeʹs Handbook of Chemistry;Science Press:Beijing,1991;p 59;translated by Shang,J.F.,Cao,S.J.,Xin,W.M.[Dean,J.A.兰氏化学手册.尚久方,操时杰,辛无名,译.北京:科学出版社,1991:59.]
(20)Gao,X.P.;Davies,J.P.;Weaver,M.J.J.Phys.Chem.1990,94(17),6858.doi:10.1021/j100380a059
(21)Moskovits,M.;Suh,J.S.J.Phys.Chem.1988,92(22),6327.doi:10.1021/j100333a030
(22)Kolega,R.R.;Schlenoff,J.B.Langmuir 1998,14(19),5469.doi:10.1021/la980553b
(23)Ji,W.;Kitahama,Y.;Han,X.X.;Xue,X.X.;Ozaki,Y.;Zhao,B.J.Phys.Chem.C 2012,116(46),24829.doi:10.1021/jp308805n
(24)Shafer-Peltier,K.E.;Haynes,C.L.;Glucksberg,M.R.;Van Duyne,R.P.J.Am.Chem.Soc.2003,125(2),588.doi:10.1021/ja028255v
(25)Amaya,R.O.;Rappoport,D.;Munoz,P.A.;Peng,P.;Mazur,E.;Guzik,A.A.J.Phys.Chem.C 2012,116(29),15568.doi:10.1021/jp302597v
(26)Le-Ru,E.C.;Blackie,E.;Meyer,M.;Etchegoin,P.G.J.Phys.Chem.C 2007,111(37),13794.doi:10.1021/jp0687908
(27)Jia,H.Y.Synthesis,Characterization of SERS Active Nanoparticles.Ph.D.Dissertation,Jilin University,Changchun,2006.[贾慧颖.银纳米粒子的制备、表征及其表面增强拉曼散射活性研究[D].长春:吉林大学,2006.]
(28)Stolberg,L.;Lipkowski,J.;Irish,D.E.J.Electroanal.Chem.1990,296,171.doi:10.1016/0022-0728(90)87241-B
(29)Trasatti,S.;Petrii,O.A.Pure&Appl.Chem.1991,63(5),711.doi:10.1351/pac199163050711
(30)Cai,W.B.;Ren,B.;Li,X.Q.;She,C.X.;Liu,F.M.;Cai,X.W.;Tian,Z.Q.Surf.Sci.1998,406,9.
(31)Álvarez-Puebla,R.A.J.Phys.Chem.Lett.2012,3(7),857.doi:10.1021/jz201625j
猜你喜欢
中国银幕(2022年4期)2022-04-07
中学生数理化(高中版.高考理化)(2021年4期)2021-07-19
分析科学学报(2021年3期)2021-07-14
色谱(2021年6期)2021-05-06
中学生数理化(高中版.高考理化)(2020年10期)2020-10-27
科技资讯(2020年12期)2020-06-03
空天防御(2020年1期)2020-04-13
表面工程与再制造(2019年6期)2019-08-24
中学数学研究(广东)(2018年23期)2018-03-05
资源节约与环保(2018年1期)2018-02-08
