
电沉积多孔复合Ni-P/LaNi5电极及其析氢电催化性能
2013-10-18 05:26段钱花王森林王丽品
物理化学学报 2013年1期
段钱花 王森林 王丽品
(华侨大学材料科学与工程学院应用化学系,福建厦门 361021)
1 引言
电催化析氢反应(HER)是电能向化学能转化的一个有效途径,对这一课题的研究,不仅对电解水制氢,而且对氯碱工业、腐蚀和阳极保护、光电化学的开发利用、化学电源及燃料电池等都具有重要意义.1,2水电解是生产高纯度氢气的重要技术,然而巨大的能源消耗妨碍了其应用,这主要是因为电解水阴极材料具有较高的析氢过电位.降低析氢反应的过电位是制氢工业中降低能耗的主要途径,而析氢反应的过电位与阴极材料的活性息息相关.因此,寻找HER优良电催化电极材料来降低能耗成为热点研究内容.3-5析氢活性的提高可通过两种方式实现:(a)增加有效的阴极表面积(几何因素);(b)降低真实的析氢过电位(电子因素).几何因素与材料的本性无关,我们可以通过各种手段增大电极的表面粗糙度.本文通过制备多孔电极,使电极的真实面积大大提高.从而提高电解过程中电极表面的真实电流密度,达到降低析氢过电位的目的.电极真实的析氢过电位是由材料的本身性能决定的,如电子功函、费米能级、金属d电子组态、表面缺陷、金属-氢化物(M-H)吸附能等,这些都能影响析氢机理与析氢反应速率,从而影响电极的析氢活性.Ni具有特殊的未成对d电子层结构,根据电催化理论,6原子氢的吸附键主要由氢原子中的电子与金属未成对的d电子形成,过渡金属原子的电子层结构中存在未成对的d电子和未充满的d轨道,可以跟吸附质(H)形成键.根据“火山型效应”,随着金属表面吸附氢键的逐渐增强,先有利于增大氢的反应速度,但若吸附过于强烈,反而会使反应速度降低.镍金属具有适中的吸附氢键能,表现出较高的反应速度,具有较好的电催化析氢性能.且镍电极制备方法简单、成本较低而受到关注.7一些研究表明,通过在电沉积镍过程中掺杂少量的P或者S能够提高镍基合金电极HER电催化活性,8,9特别是磷质量分数为6%-9%的非晶态Ni-P合金活性高.10非晶态材料与晶态材料不同,具有长程无序、短程有序的原子结构.致密、无序的原子堆积形式及所具有的空间结构使其表现出较高的化学活性.另外,非晶态合金的表面自由能较高,处于亚稳状态,这种亚稳态结构能有效地降低氢原子在金属表面吸附的活化能,这些都使非晶态合金较之同成分的晶态材料具有更高的催化活性.同时非晶态Ni-P合金镀层还具有高的机械强度和优越的耐蚀性,传统上用于表面精饰、防腐和抗磨等.11Ordine等12研究了镀液pH值对Ni-P电沉积的影响及其沉积机理.近年来,具有吸/放氢能力的储氢合金,尤其是AB5型储氢合金,用作电解水析氢阴极受到了较大的关注.13金属间化合物LaNi5具有六方结构,其中有许多间隙位置,可以固溶大量的氢,在电解过程中实现储氢功能,而在断电时这些吸附氢又可以在阴极发生放电反应,防止电极被空气氧化和腐蚀,从而起到保护电极电催化活性的作用(即电极具有良好的抗断电性能).14,15通过复合电沉积可以将一种或数种不溶性固体微粒掺杂到金属材料中形成复合镀层,通过几种材料的正协同效应得到有效的电催化活性表面.同时由于稀土元素具有独特的4f层电子结构,使稀土金属及其合金具有较高的催化活性.16Tanaka等17报道了LaNi5合金的表观交换电流密度对数值(lg jo)达到-3.5 A·cm-2,非常接近贵金属Pd(-3.9 A·cm-2)和Pt(-3.5 A·cm-2)的值.但由于氢气的吸、脱附过程容易导致储氢材料体积的交替变化产生碎裂,而无法单独使用.因此可以通过复合电沉积的技术将这些高催化活性的固体颗粒复合到金属或合金基体中去,形成具有高比表面积和高催化活性的复合电极材料.本文先通过一步复合电沉积将LaNi5和Al颗粒囊嵌到镀层中得到了Ni-P/(LaNi5+Al)复合镀层,然后采用碱溶法将镀层中的铝溶解掉制得多孔复合Ni-P/LaNi5电极.采用扫描电镜和X射线衍射等分析技术表征了电极结构,运用电化学技术研究了电极的析氢电催化性能及电化学稳定性并与相应的多孔Ni-P电极进行对比.
2 实验部分
2.1 电极制备
电沉积镍-磷溶液组成及工艺条件:NiSO4·6H2O(250 g·L-1);NiCl2·6H2O(50 g·L-1);NaH2PO2(20 g·L-1);H3BO3(40 g·L-1);电流密度为50 mA·cm-2,镀液pH值为4.0-4.5,温度为45°C,时间为20 min.所用试剂均为分析纯,镀液由一次蒸馏水配制.阴极材料为1.7 cm×1.7 cm×0.2 cm的黄铜片,阳极为6 cm×9 cm×0.4 cm的纯镍板,将阴极和阳极平行置于镀液中.
固体微粒预处理.铝粉30 g·L-1(平均粒径10 μm),LaNi525 g·L-1(平均粒径20 μm,厦门金鹭有限公司).将这些固体粉末混合放入pH值为8.0的弱碱中超声除油、过滤,用蒸馏水冲洗干净,再放入1 g·L-1的聚乙二醇中充分搅拌1 h,过滤,用蒸馏水洗净,烘干.18
复合电极制备.将处理好的固体微粒倒入Ni-P镀液中,控制电镀液pH值在4.0-4.5,充分搅拌(磁力搅拌)直至固体粉末均匀地分散在镀液中,然后再进行电镀,在加入铝粉的镀液中制备Ni-P/Al复合镀层,在加入LaNi5和铝粉的镀液中制备Ni-P/(LaNi5+Al)复合镀层.将得到的Ni-P/Al复合镀层和Ni-P/(LaNi5+Al)复合镀层放置于60°C的6 mol·L-1NaOH溶液中除铝,待无气泡冒出,将其取出,用蒸馏水清洗,吹干分别得到多孔Ni-P电极和多孔复合Ni-P/LaNi5电极.
2.2 电极结构及性能测试
用日本Hitachi公司生产的S-3500N扫描电镜(SEM)观察电极的表面形貌,镀层成分分析用该扫描电镜附带的ISIS-300能谱仪(英国牛津公司)测定.结构分析在Panalytical Xʹpert PRO粉末X射线衍射仪(XRD)(荷兰PANalytical公司)上进行,Cu Kα射线,测试所用基体为1.7 cm×1.7 cm×0.2 cm的黄铜片,而镀层组成和表面形貌测试所用基体为1.7 cm×1.7 cm×0.05 cm的紫铜片.
利用CHI-630D电化学综合测定仪(上海辰华仪器公司)进行电化学测试,包括开路电位、阴极极化、循环伏安、恒电位间断及长时间电解.利用Parstat 2273电化学工作站(美国Princeton Applied Research公司)进行电化学阻抗测试,频率范围:100 kHz-0.01 Hz,E=5 mV.采用三电极体系进行测量,使用玻璃三室电解槽(150 mL).工作电极为自制的多孔复合电极,辅助电极为3 cm×3 cm大面积铂电极,参比电极选用Hg/HgO电极(10%(w)NaOH),电解液为20%(w)NaOH溶液.
3 结果与讨论
3.1 镀层的表面形貌及结构

图1 不同镀层的扫描电子显微镜图Fig.1 SEM images of different coatings
图1示出不同镀层的SEM图,均放大1000倍.图1(a)为Ni-P合金镀层,可以看出镀层相对比较平整.图1(b)为Ni-P/Al的复合镀层,可以看到呈白色物质Al颗粒较均匀囊嵌在镀层中.图1(c)为多孔Ni-P镀层,是由复合Ni-P/Al电极在60 °C、6 mol·L-1的NaOH溶液中通过浸泡除铝得到的.可以观察到镀层表面分布着很多大小不均匀的小孔,这些孔洞大大增加了镀层的比表面积.图1(d)为多孔复合Ni-P/LaNi5镀层,它是由Ni-P/(LaNi5+Al)镀层在60°C,6 mol·L-1的NaOH溶液中通过浸泡除去铝粉得到的.LaNi5颗粒较均匀囊嵌在镀层中,且镀层表面分布着许多小孔.多孔复合Ni-P/LaNi5镀层的能谱图如图2所示,显然镀层的成分是Ni、P和La,谱图中未发现其他的元素.

图2 多孔复合Ni-P/LaNi5镀层的EDS能谱图Fig.2 EDS spectrum of the porous composite Ni-P/LaNi5coating
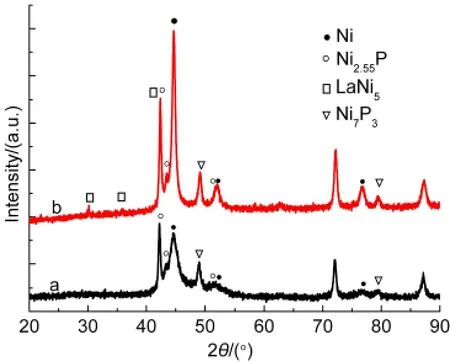
图3 不同镀层的XRD谱图Fig.3 XRD patterns of different coatings
图3是不同镀层的XRD谱图.曲线a对应的是多孔Ni-P镀层,可以看出多孔Ni-P镀层中Ni的衍射峰比较弱,分别出现在 2θ=44.487°,51.859°,76.378°处,空间晶形为面心立方(Fm3m),对应PDF卡号为65-2865.曲线b对应的是多孔复合Ni-P/LaNi5镀层,在 2θ=30.204°,35.848°,42.308°的位置出现了LaNi5的衍射峰,空间晶形为六方密堆积(P6/mmm),对应 PDF 卡号为 65-0093.在 2θ=44.601°,51.917°,76.778°的位置同样出现了Ni的衍射峰.从两样品的XRD谱图可以看出,两图谱均出现了馒头峰,说明随着P的加入,Ni和P的结构由晶体逐渐转化为晶体和非晶体的混合形式.19且随着LaNi5颗粒的引入,得到的多孔复合Ni-P/LaNi5的衍射峰有所增强,表明多孔复合Ni-P/LaNi5电极的结晶度较多孔Ni-P电极强.以上两XRD谱图都没有出现P的衍射峰,通过EDS测试得到多孔Ni-P镀层中各元素原子分数分别为Ni 90.74%,P 9.26%.多孔Ni-P/LaNi5镀层中各元素原子百分含量分别为Ni 94.43%,P 3.07%,La 2.5%,由La的含量可以推出多孔复合Ni-P/LaNi5镀层中LaNi5相中的Ni占12.5%,故Ni-P合金相中Ni占81.93%;P占3.07%.通过软件分析,推测在42.266°,43.287°,51.514°的位置可能出现了Ni2.55P的衍射峰,在49.074°和79.541°可能出现了Ni7P3的衍射峰,在电沉积过程中Ni和P可能形成了金属间化合物.
3.2 电极的HER电催化性能
图4示出多孔Ni-P电极和多孔复合Ni-P/LaNi5电极常温下的阴极极化曲线.图5为多孔复合Ni-P/LaNi5电极在不同温度下的阴极极化曲线.扫描范围为从开路电位到-1.3 V(vs Hg/HgO,以下相同),扫描速率为0.001 V·s-1.由图4可以得出,多孔复合Ni-P/LaNi5电极在约-0.820 V有一个还原峰,可能发生原子氢吸附,在-0.975 V左右开始大量析氢,而多孔Ni-P电极在-1.055 V左右才开始大量析氢,与多孔Ni-P电极相比,多孔复合Ni-P/LaNi5电极具有更正的起始析氢电位.

图4 不同电极的阴极极化曲线Fig.4 Cathodic polarization curves of different electrodes
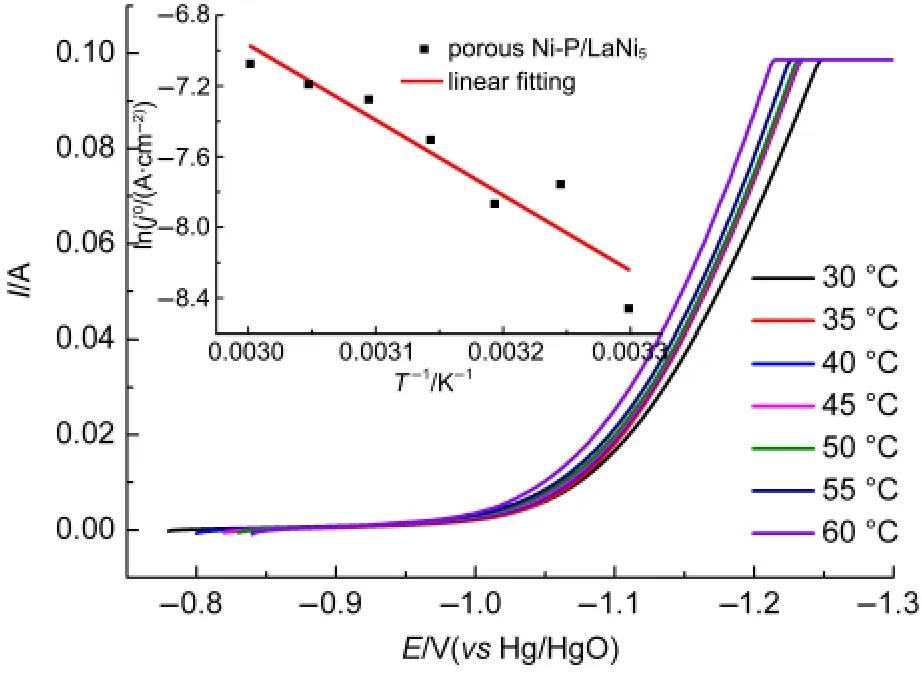
图5 多孔复合Ni-P/LaNi5不同温度的阴极极化曲线Fig.5 Cathodic polarization curves of different temperatures of the porous composite Ni-P/LaNi5
从图5中可以看出,随着温度的升高,电极的起始析氢电位有较明显的正移,说明析氢电催化性能与温度有关,温度越高,电催化活性越强.这是由于温度越高,分子运动越剧烈,加速了电荷的转移以及离子的移动.20根据阴极极化曲线中在极化较大时的数据,做η-ln j的Tafel曲线,以电极的开路电位近似作为体系的平衡电位.根据Tafel关系式:η=a+b ln j,当阴极极化时,a=-(RT/αnF)ln j0,b=RT/αnF,式中η为过电位(η=Eocp-E),j为电流密度,T为温度,R为气体常数,n为转移电子数,F为法拉第常数,α为传递系数.通过Tafel曲线线性拟合,从而得到析氢过程的表观交换电流密度j0,而j0=Fkαexp(-Ea/RT),其中k为常数,α为传递系数,Ea为表观活化自由能.通过测量不同温度下的交换电流密度j0,将ln j0对1/T作图则有:d ln j0/d(1/T)=-Ea/R,如图5中的插图(为多孔复合Ni-P/LaNi5电极).可以得到电极析氢反应的表观活化能Ea数据,见表1.显然,随着温度的升高,多孔Ni-P电极和多孔复合Ni-P/LaNi5电极的交换电流密度都不断增强,也证明了电极的析氢电催化活性随着温度的升高而增强.计算得到多孔复合Ni-P/LaNi5电极的表观活化能为35.44 kJ·mol-1,比多孔Ni-P电极表观活化能(50.91 kJ·mol-1)要低.这也说明多孔复合Ni-P/LaNi5电极比多孔Ni-P电极具有更好的电催化析氢性能.

表1 不同电极的电化学析氢动力学参数Table 1 Kinetic parameters for electrochemical hydrogen evolution reaction of different electrodes
图6是多孔Ni-P电极和多孔复合Ni-P/LaNi5电极常温下的循环伏安曲线.电位扫描范围为从开路电位到-1.3 V,扫描速度均为0.01 V·s-1.当电位从开路电位开始往负扫时,多孔复合Ni-P/LaNi5电极在-0.787 V时出现了一个还原峰,可能是由于LaNi5具有很强的吸附氢的能力,在此时发生了氢的吸附.在-0.950 V时开始明显析氢,而多孔Ni-P电极在-1.085 V才开始出现明显析氢.当电位从-1.3 V往正扫描时,多孔Ni-P电极分别在-0.910和-0.606 V时出现了氧化峰,对应吸附氢脱附氧化和Ni-P氧化峰.而多孔复合Ni-P/LaNi5电极没有出现明显的氧化峰,但是有阳极氧化电流,可能是储氢合金在阴极过程储存的吸附大量氢不断氧化,防止了Ni-P被氧化.也可以看出多孔复合Ni-P/LaNi5电极比多孔Ni-P电极具有更正的起始析氢电位,这与阴极极化曲线得出的结论一致.

图6 不同电极的循环伏安曲线Fig.6 Cyclic voltammetric curves of different electrodes
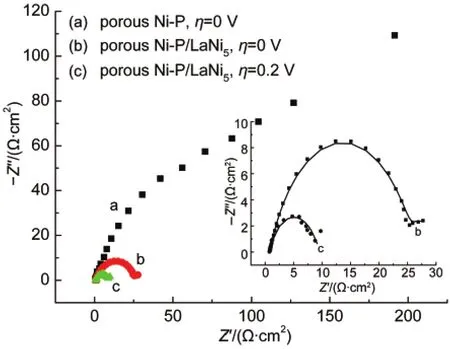
图7 不同电极的电化学阻抗图Fig.7 Nyguist plots of different electrodes
图7显示多孔Ni-P电极和多孔复合Ni-P/LaNi5电极常温下的电化学阻抗图,其中插图为多孔复合Ni-P/LaNi5电极在无偏压(η=0 V)和-0.2 V偏压(η=0.2 V)下的电化学阻抗的拟合图.图8为相应的拟合电路的等效电路,拟合得到的参数值见表2.在等效电路及表中,Rsl为溶液电阻,Rct为电荷转移电阻(反应(1)),Cdl为电极表面的双电层电容,RW为电化学脱附电阻(即极化电阻)(反应(2)),Q为CPE,是常相位元件,表示与电容的相似程度,CPE值越大表示电极表面越光滑,电极真实表面积越小.21图7曲线(a)为多孔Ni-P在开路电位下的Nyquist图,可以看出它为一段半径较大的圆弧,可近似为直线,是典型的电荷转移电阻和双电层电容并联.22曲线(b)、(c)分别为多孔Ni-P/LaNi5电极在开路电位和-0.2 V偏压下的Nyquist图,可以看出在高频区由半圆组成,说明发生电化学反应,在低频区为短斜线,这反映了其Warburg阻抗特征,说明电极反应机理是由扩散控制步骤,即浓差极化控制.23由(a)和(b)曲线可以看出它们的圆弧半径逐渐减少,说明随着LaNi5颗粒的加入,复合电极的析氢反应总电阻有较大的减小,反映出其析氢过电位有所降低.由(b)和(c)曲线可以看出,当在电极两端加有负偏压时,其电阻也变小,通过表2的拟合参数也可以看出Rct和Rw电阻都有所减少.碱性介质HER机理如下:24,25
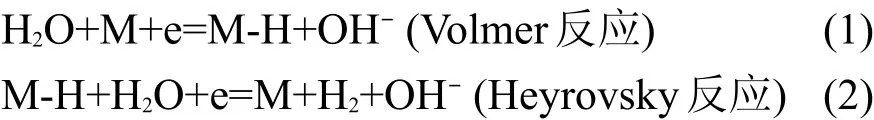
一般地,在该电极析氢电位下,控制步骤为电化学脱附或电化学脱附和氢气扩散联合过程.

图8 多孔Ni-P电极和多孔复合Ni-P/LaNi5电极的等效电路图Fig.8 Electrial equivalent circuit used for simulating the impedance spectra of porous Ni-P electrode and porous composite Ni-P/LaNi5electrode

表2 多孔Ni-P电极和多孔复合Ni-P/LaNi5电极析氢反应等效电路的拟合参数值Table 2 Fitted parameter values of eletrical equivalent circuit of the porous Ni-P electrode and porous composite Ni-P/LaNi5electrode

图9 多孔复合Ni-P/LaNi5电极在不同偏压下的电化学阻抗图Fig.9 Nyquist plots of the porous composite Ni-P/LaNi5at different bias voltages
图9为多孔复合Ni-P/LaNi5电极常温下在不同偏压下的电化学阻抗图.可以看出在高频区都均为半圆弧(电化学步骤控制),低频近似为直线(扩散步骤控制),且随着偏压增大,圆弧半径逐渐减小,说明电化学反应电阻随着偏置电压的增大而减小.26
对多孔Ni-P电极和多孔复合Ni-P/LaNi5电极的Nyquist图进行拟合,得到其表面的双电层电容分别为5137和68300 μF·cm-2.已知光滑固体电极的双电层电容值为60 μF·cm-2,由此可计算出电极的电化学比表面积(Ss),则真实表面积(Sr),Sr=Cdl/60,和粗糙度r=Sr/Ss,计算结果如表3所示,可以看出多孔复合Ni-P/LaNi5电极的比表面积为1138.3 cm2,远远大于多孔Ni-P电极的85.7 cm2.说明溶液电阻和极化电阻与电极的真实表面积密切相关,当电极的真实表面积增大时,其电阻有较明显的减少,电极的电催化活性有了较大的提高.
3.3 电极的电解HER稳定性

表3 不同电极的表面参数Table 3 Surface parameters of different electrodes
图10示出多孔Ni-P电极和多孔复合Ni-P/LaNi5电极(两电极表观面积相同)常温下在20%(w)NaOH溶液中的恒电位间断电解曲线.测试条件是在V=-1.3 V下先电解2 h后间断1 h,多次循环,在第7次电解2 h间歇为8 h,后面仍为电解2 h间歇1 h,直到共间断电解10次结束.从图中可以看出,多孔Ni-P电极前几次电解得到的曲线不够稳定,随着电解次数的增加,表观电流有增大的趋势,后又趋于减小;而多孔Ni-P/LaNi5电极经过10次断电电解前后曲线比较稳定,电流基本维持在-0.0984 A附近.说明了相对于多孔Ni-P电极具有更高的抗断电稳定性,这主要是因为LaNi5具有较强的吸附氢的能力,在断电时吸附的氢又释放出来,有效抑制电极表面被氧化,从而起到了保护电极的作用.27
为了进一步研究多孔Ni-P/LaNi5电极的稳定性,我们将多孔Ni-P电极和多孔复合Ni-P/LaNi5电极分别在常温下进行恒电位长时间电解,控制电位V=-1.3 V,电解时间为20 h.由图11曲线a可以看出,多孔Ni-P开始比较稳定,后又有波动电流变大再趋于稳定,电流在-0.01 A上下波动;由曲线(b)可以看出多孔Ni-P/LaNi5电极的电流较多孔Ni-P高且曲线平稳,基本维持在-0.0984 A附近.这说明多孔Ni-P/LaNi5电极具有更高的稳定性和电化学活性,和图10得到的结论一致.

图10 不同电极的恒电位间断电解Fig.10 Constant potential with discontinuity electrolysis of different electrodes

图11 不同电极的恒电位长时间电解Fig.11 Constant potential electrolysis of different electrodes
3.4 电极稳定性能原因探讨
图12为多孔复合Ni-P/LaNi5电极常温下的恒电位长时间电解后的开路电位随时间的变化,测试时间为30 min.由图可见,该电极的开路电位逐渐正移,随着时间的进行正移速度比较缓慢,半小时后,开路电位仍保持在-0.942 V,这比电解前开路电位(-0.647 V)还要负,因为在电解的过程中电极不断析氢,使得在电解停止后该电极中LaNi5吸附了大量的原子氢而达到了饱和,这些吸附氢在电极断电后不断释放,因此电极的开路电位不断正移.且由于电极中LaNi5吸附了大量氢,在断电后不断释放并与空气中的氧气结合,阻止了电极金属相物质被空气氧化和腐蚀,维持了电极的电催化活性,因此具有一定的抗断电性能.28

图12 多孔复合Ni-P/LaNi5电极的恒电位长时间电解后的开路电位曲线Fig.12 Open-circuit potential(OCP)curves after constant potential electrolysis of porous composite Ni-P/LaNi5electrode

图13 多孔Ni-P/LaNi5的阳极极化曲线Fig.13 Anodic polarization curves of the porous composite Ni-P/LaNi5electrode
为进一步测试该电极吸附氢的量,研究了多孔复合Ni-P/LaNi5常温下不饱和吸附氢和饱和吸附氢的阳极极化曲线.如图13所示,曲线a为该电极不饱和吸附氢(制备态电解前该电极在制备的过程中也吸附了一定的氢)的阳极极化曲线,其开路电位为-0.647 V,阳极极化范围为-0.65-0.25 V,再将该电极在恒电位(-1.3 V)下电解30 min得到饱和吸附氢电极,曲线b为该电极饱和吸附氢后的阳极极化曲线,开路电位为-0.830 V,阳极极化范围为-0.83-0.25 V,扫描速率均为0.002 V·s-1,从图中可以看出不饱和吸附氢电极在-0.498 V时出现了第一个氧化峰,在-0.039 V时出现了第二个氧化峰,饱和吸附氢电极在-0.384 V时出现了第一个氧化峰,在-0.015 V时出现了第二个氧化峰(Ni电极表面的氧化).29第一氧化峰即为该电极吸附氢的氧化造成,从图中可以看出饱和吸附氢电极的氧化峰电流远大于不饱和吸附氢电极,这是由于电解后该电极LaNi5吸附的氢达到饱和,吸附在电极中的氢的量多.电解前该电极在制备的过程中也吸附了一定的原子氢但没有达到饱和.
4 结论
(1)运用一步复合电沉积制备了Ni-P/Al和复合Ni-P/(LaNi5+Al)镀层,通过浓碱浸泡除铝分别得到了多孔Ni-P和多孔复合Ni-P/LaNi5镀层;(2)多孔复合Ni-P/LaNi5电极比多孔Ni-P电极具有更低的析氢过电位、更高的比表面积和更高的电催化活性;(3)多孔复合Ni-P/LaNi5电极具有优异的抗断电稳定性.
(1)Wei,Z.D.;Yan,A.Z.;Feng,Y.C.;Li,L.;Sun,C.X.;Shao,Z.G.;Shen,P.K.Electrochem.Commun.2007,9,2709.doi:10.1016/j.elecom.2007.09.006
(2)Abdel-Karim,R.;Halim,J.;El-Raghy,S.;Nabil,M.;Waheed,A.J.Alloy.Compd.2012,530,85.doi:10.1016/j.jallcom.2012.03.063
(3)Suffredini,H.B.;Cerne,J.L.;Crnkovic,F.C.;Machado,S.A.S.;Avaca,L.A.Int.J.Hydrog.Energy 2000,25,415.doi:10.1016/S0360-3199(99)00049-X
(4)Solmaz,R.;Gündoğdu,A.;Döner.A.;Kardas,G.Int.J.Hydrog.Energy 2012,37,8917.doi:10.1016/j.ijhydene.2012.03.008
(5)Esposito,D.V.;Hunt,S.T.;Kimmel,Y.C.;Chen,J.G.J.Am.Chem.Soc.2012,134,3025.doi:10.1021/ja208656v
(6)Jakšić,J.M.;Vojnović,M.V.;Krstajić,N.V.Electrochim.Acta 2000,45,4151.doi:10.1016/S0013-4686(00)00549-1
(7)Han,Q.;Wei,X.J.;Liu,K.R.The Chinese Journal of Nonferrous Metals 2001,11(1),158.[韩 庆,魏绪均,刘奎仁.中国有色金属学报,2001,11(1),158.]
(8)Kim,H.S.;Lee,H.;Han,K.S.;Kim,J.H.;Song,M.S.;Park,M.S.;Lee,J.Y.;Kang,J.K.J.Phys.Chem.B 2005,109,8983.
(9)Fetohi,A.E.;Abdel Hameed,R.M.;El-Khatib,K.M.;Souaya,E.R.Int.J.Hydrog.Energy 2012,37(9),7677.doi:10.1016/j.ijhydene.2012.01.145
(10)Paseka,I.Electrochim.Acta 1995,40(11),1633.doi:10.1016/0013-4686(95)00077-R
(11)Chen,T.Y.Composite Electroplating Ni and Special Electro-Plating Ni,1st ed.;Chemical Industry Press:Beijing,2008;pp 18-71.[陈天玉.复合镀镍和特种镀镍.北京:化学工业出版社,2008:18-71.]
(12)Ordine,A.P.;Díaz,S.L.;Margarit,I.C.P.;Barcia,O.E.;Mattos,O.R.Electrochim.Acta 2006,51,1480.doi:10.1016/j.electacta.2005.02.129
(13)Srivastava,S.;Upadhyaya,R.K.Int.J.Hydrog.Energy 2011,36,7114.doi:10.1016/j.ijhydene.2011.02.111
(14)Xu,Y.H.;He,G.R.;Wang,X.L.Int.J.Hydrog.Energy 2003,28,961.doi:10.1016/S0360-3199(02)00170-2
(15)Cuscueta,D.J.;Corso,H.L.;Arenillas,A.;Martinez,P.S.;Ghilarducci,A.A.;Salva,H.R.Int.J.Hydrog.Energy 2012,37,14978.doi:10.1016/j.ijhydene.2011.12.101
(16)Natalia,C.;Tebello,N.Electrochim.Acta 1997,42,3519.doi:10.1016/S0013-4686(97)00033-9
(17)Tanaka,K.;Okazaki,S.;Ichitsubo,T.;Yamamoto,T.;Inui,H.;Yamaguchi,M.;Koiwa,M.Intermetallics 2000,8,613.doi:10.1016/S0966-9795(99)00154-5
(18)Wang,S.L.;Zhang,Y.Acta Phys.-Chim.Sin.2011,27(6),1417.[王森林,张 艺.物理化学学报,2011,27(6),1417.]doi:10.3866/PKU.WHXB20110510
(19)Cao,Y.L.;Wang,F.;Liu,J.J.;Wang,J.J.;Zhang,L.H.;Qin,S.Y.Acta Phys.-Chim.Sin.2009,25(10),1979.[曹寅亮,王 峰,刘景军,王建军,张良虎,覃事永.物理化学学报,2009,25(10),1979.]doi:10.3866/PKU.WHXB20091017
(20)Liu,Y.;Her,J.H.;Dailly,A.;Ramirez-Cuesta,A.J.;Neumann,D.A.;Brown,C.M.J.Am.Chem.Soc.2008,130,11813.doi:10.1021/ja803669w
(21)Norlin,A.;Pan,J.;Leygraf,C.Biochem.Eng.J.2002,19,67.
(22)Herraiz-Cardona,I.;Ortega,E.;Antón,J.G.;Pérez-Herranz.Int.J.Hydrog.Energy 2011,36,9428.doi:10.1016/j.ijhydene.2011.05.047
(23)Zhang,J.Q.Electrochemical Measurement Technology,1st ed.;Chemical Industry Press:Beijing,2010;pp 204-237.[张鉴清.电化学测试技术.北京:化学工业出版社,2010:204-237.]
(24)Baba,R.;Nakabayashi,S.;Fujishima,A.J.Phys.Chem.1985,89(10),1902.doi:10.1021/j100256a018
(25)Mihailov,L.;Spassov,T.;Bojinov,M.Int.J.Hydrog.Energy 2012,37(14),10499.
(26)Bao,J.Z.;Wang,S.L.Acta Phys.-Chim.Sin.2011,27(12),2849.[鲍晋珍,王森林.物理化学学报,2011,27(12),2849.]doi:10.3866/PKU.WHXB20112849
(27)Yang,S.Q.;Han,S.M.;Song,J.Z.;Li,Y.Journal of Rare Earths 2011,29(7),692.doi:10.1016/S1002-0721(10)60524-8
(28)Han,Q.;Chen,J.S.;Liu,K.R.;Wei,X.J.Acta Metal.Sin.2008,744,887.[韩 庆,陈建设,刘奎仁,魏绪钧.金属学报,2008,744,887.]
(29)Figueroa-Torres,M.Z.;Domínguez-Ríos,C.;Cabaňas-Moreno,J.G.;Vega-Becerra,O.Int.J.Hydrog.Energy 2012,37,10743.doi:10.1016/j.ijhydene.2012.04.097
猜你喜欢
金属热处理(2022年10期)2022-10-25
电镀与精饰(2022年8期)2022-08-18
建材发展导向(2021年7期)2021-07-16
青年歌声(2019年2期)2019-02-21
中国有色金属学报(2018年2期)2018-03-26
机械工程师(2018年10期)2018-02-18
知识经济·中国直销(2017年11期)2017-11-28
中国公路(2017年16期)2017-10-14
环境保护与循环经济(2017年1期)2017-09-26
中南大学学报(自然科学版)(2016年2期)2017-01-19
