
Bi掺杂锐钛矿相TiO2第一性原理计算
2013-10-17 03:02:52吴国浩郑树凯
无机化学学报 2013年1期
吴国浩 郑树凯*, 吕 霄
(1河北大学电子信息工程学院,保定 071002)
(2河北大学计算材料研究中心,保定 071002)
(3清华大学材料科学与工程系,北京 100084)
锐钛矿相TiO2是一种Ⅳ-Ⅵ族间接宽带隙氧化物半导体光催化材料,室温下禁带宽度为3.2 eV,与其它半导体光催化剂相比,具有良好的化学稳定性、低成本、制备简单和无毒等优点,在废水和废气的净化处理方面有着广阔的应用前景,引起了广大科研工作者对其广泛的研究[1-7]。但较大的禁带宽度导致其只能被占太阳光总能量4%,波长小于或等于387.5 nm的紫外光激发,严重限制了其在光电催化领域的大规模应用。因此如何通过改性提高其光谱吸收范围和光催化活性是锐钛矿相TiO2大规模应用的关键。通过掺杂对TiO2进行带隙调控,从而获取可以响应可见光的TiO2基催化剂一直是人们努力的目标。如Ding等[8]采用溶胶-凝胶法制备了Co掺杂锐钛矿相TiO2,紫外-可见吸收光谱测量发现,掺杂后TiO2吸收出现明显红移,并在600 nm左右出现一个额外吸收峰;Gao等[9]采用固-液反应法制备了S掺杂TiO2,在可见光下通过对罗丹明B的降解发现,S掺杂TiO2降解率达到97.9%,是未掺杂TiO2的6倍;Deng等[10]制备了Mn掺杂锐钛矿相TiO2纳米粉,通过对亚甲基蓝的降解发现Mn掺杂提高了TiO2催化活性,并且Mn掺杂后TiO2出现明显的吸收带边红移;Zhang等[11]采用水热法制备了Ⅰ掺杂TiO2纳米粉,通过对CO2的光致还原发现,Ⅰ掺杂TiO2相对于未掺杂TiO2在可见光和紫外光下都出现明显增强。
近年来,实验上对Bi掺杂TiO2光催化活性的研究也非常活跃。如Huang等[12]采用超声雾化热解法合成了不同Bi掺杂浓度的TiO2,通过对甲基橙的降解发现,在Bi掺杂浓度为2%时,TiO2光催化效果最好;Rengaraj等[13]通过对Bi3+掺杂TiO2光降解甲基对硫磷的研究发现,掺杂后光降解效率大大提高;梁晓明等[14]采用低温醇解法制备了Bi掺杂TiO2光催化剂,发现掺杂后TiO2在紫外和太阳光下光催化活性明显提高;Xu等[15]采用金属有机分解法(MOD)制备了Bi掺杂锐钛矿相TiO2,通过对甲基橙的分解,发现对比于未掺杂TiO2,掺杂后电子-空穴复合受到抑制,同时催化活性大大提高,并观察到吸收光谱的红移。由于实验过程中采用的工艺条件各不相同,而影响TiO2光催化活性的因素又非常复杂,同时由于缺少Bi元素对TiO2电子结构影响的详细研究,从而导致对掺杂改性的机理说法不一。据我们所知,目前关于Bi掺杂TiO2的理论研究较少。因此本文采用基于密度泛函理论下的第一性原理平面波超软赝势方法计算分析了Bi掺杂前后锐钛矿相TiO2的电子结构和光学性质,从量子化学角度对掺杂后锐钛矿型Ti02光催化活性增强的机理进行了探讨,以期为实验和实际应用提供相关理论基础。
1 模型和计算方法
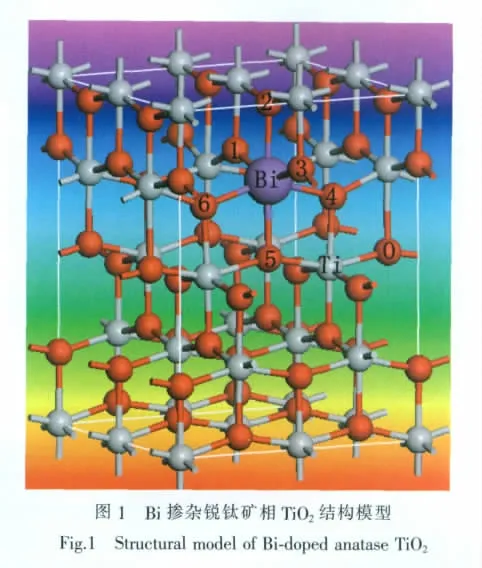
锐钛矿相TiO2属于正方晶系结构,其空间点群为I41/amd(D4h19),其中每个八面体与周围8个八面体相连接 (4个共边,4个共角),4个TiO2分子构成一个晶胞,每个基本晶胞内包含12个原子。如图1所示,本文在2×2×1 TiO2超晶胞结构中用Bi替代图中位置的Ti(图中Bi的大小不是Bi的实际大小,目的是为突出Bi原子),建立了Ti15BiO32超晶胞,原子个数为48,掺杂后Bi的掺杂浓度为1.56at%比较符合实际掺杂[13]。
本文采用Materials Studio软件中的量子力学模块Castep对掺杂前后锐钛矿相TiO2的相关性质进行了计算。计算时采用非自旋极化进行处理,计算中首先对超晶胞进行结构优化,优化中电子间相互作用的交换关联能由广义梯度近似(GGA)下的PBE进行描述,能量收敛标准设为 5×10-7eV·atom-1,平面波截止能设为400 eV,第一布里渊区按7×7×6进行分格,自洽收敛能精度平均每个原子为5.0×10-7eV·atom-1,最大位移为5.0×10-5nm,晶体内应力收敛标准为0.02 GPa,原子间相互作用力收敛标准为0.01 eV·nm-1,然后对能带结构、态密度(包括分态密度)、电荷布居和光学性质进行了计算 (计算时条件不变),参与计算的价电子为 Ti:3s23p63d24s2,O:2s22p4,Bi:6s26p3,所有计算都在倒易空间中进行。
2 结果与分析
2.1 几何优化结果
对掺杂前后TiO2模型进行几何优化后,晶格产生畸变,晶格常数发生变化,表1给出了不同情况下晶格常数值,对比未掺杂锐钛矿相TiO2发现,Bi掺杂后TiO2晶格常数和体积都有不同程度的变大。这从三方面可以解释:①对于Bi掺杂来说主要是因为Bi3+的离子半径 (103 pm)远大于Ti4+的离子半径(60.5 pm);②Bi-O键长大于Ti-O键长,(Ti-O键长为0.220 5、0.194 6和0.194 6 nm,而Bi-O对应键长为0.222 0、0.216 0 和 0.215 9 nm);③ Bi3+掺杂 TiO2取代晶体中的Ti4+,晶体中缺少1个电子,使得多余正电荷之间相互排斥作用增大,引起体积增大。
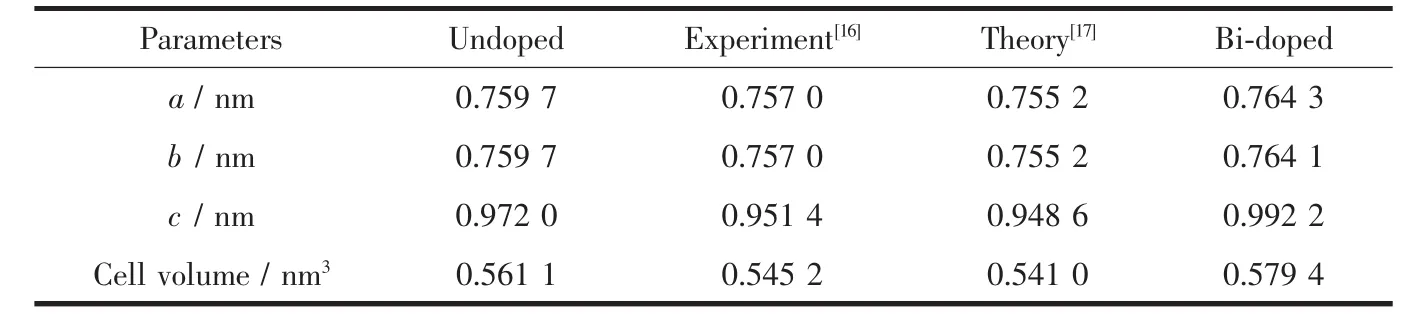
表1 Bi掺杂前后锐钛矿相TiO2晶格常数值和体积对比Table 1 Geometry optimized structural parameters and volume of the pure anatase TiO2crystal comparing with the Bi-doped TiO2
2.2 电荷布居
通过电荷布居分析可以知道各原子轨道上的电子分布,进而确定原子间的成键情况。表2和表3是Bi掺杂前后锐钛矿相TiO2的电荷布居表,超晶胞内凡是O原子或Ti原子电荷布居数相同的仅列出一组,表3中加粗部分为Bi原子周围的O原子,由于对称,所以只列出3个,如图1中所示,而所有Ti原子都受到影响,电荷布居数均有变化。对比表2和表3发现,取代Ti位掺杂的Bi离子的电荷布居数为2.37。 掺杂前O原子电荷布居数为-0.67,掺杂后临近Bi的O的电荷布居都有所数增大;掺杂前Ti的电荷布居数为1.33,掺杂后Ti的电荷布居数都有所下降。表明Bi掺杂后O原子周围的电负性增加,Ti原子周围的电正性有所减弱,说明掺杂后共价键减弱,离子键增强。
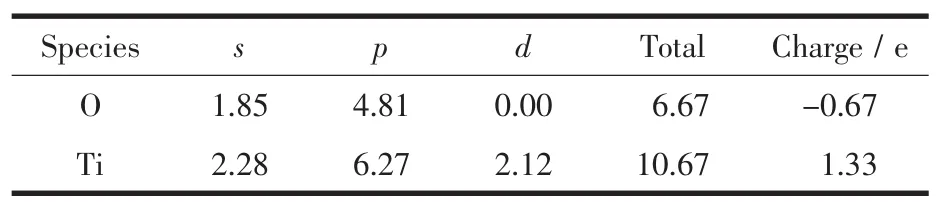
表2 纯锐钛矿相TiO2超晶胞中电荷布居数Table 2 Atomic populations of pure anatase TiO2supercell
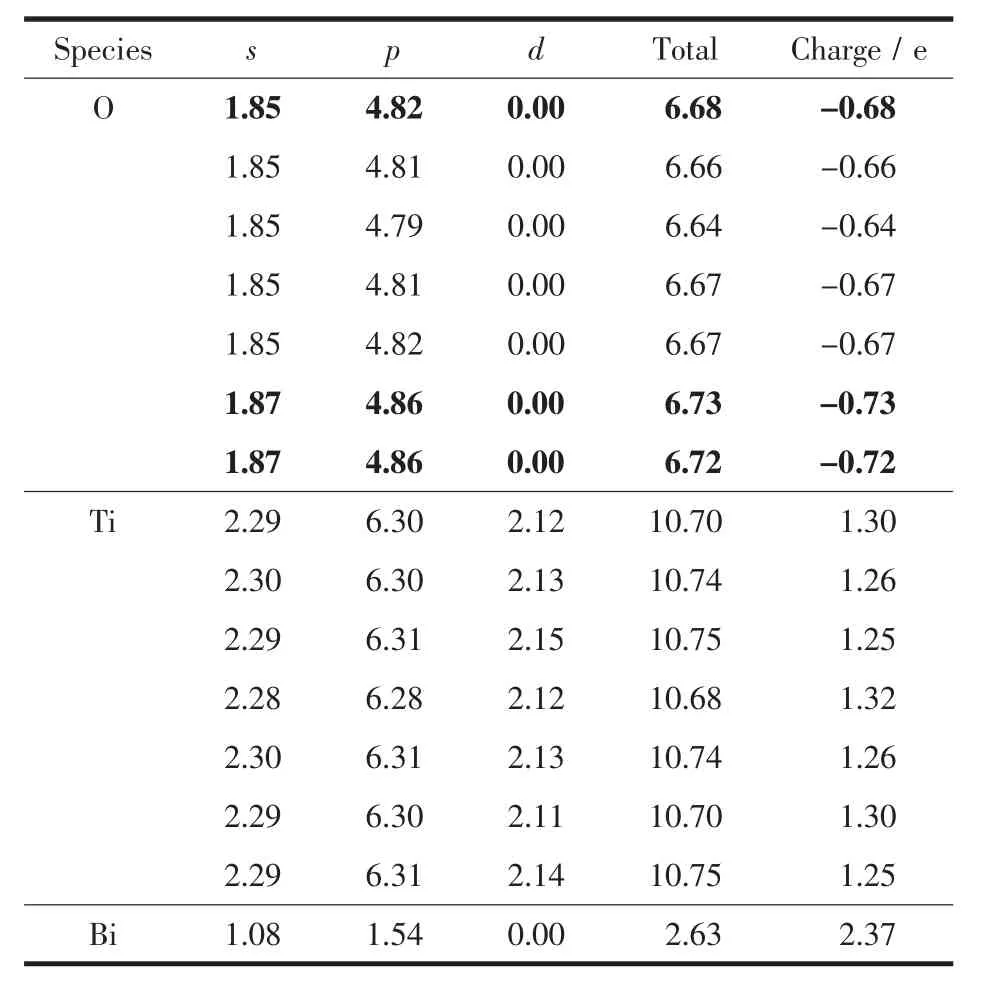
表3 掺杂Bi后锐钛矿相TiO2超晶胞中电荷布居数Table 3 Atomic populations of Bi-doped TiO2 supercell
2.3 能带结构
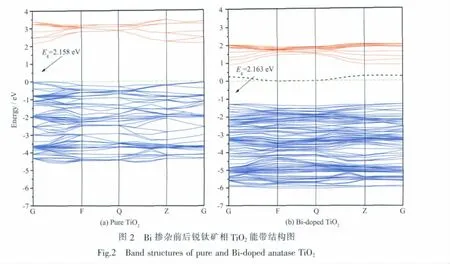
禁带宽度是影响半导体光催化的一个重要因素。图2的(a~b)分别是锐钛矿相TiO2掺杂前后沿第一布里渊区高对称方向的能带结构图,能带图中始终将电子能够填充的最高能级作为能量零点 (图b中靠近能量零点的黑色虚线为杂质能级)。由图2(a)可知纯锐钛矿相TiO2禁带宽度为2.158 eV,小于锐钛矿相TiO2实验值3.23 eV,这是由于GGA近似带来的影响。在采用广义梯度近似(GGA)处理一些体系时,由于交换关联能只计入某处的电荷密度对交换关联能的影响,并不能完全描述真实的多电子相互作用能,通常会低估带隙,这在相关文献中[18-19]已有详细讨论。但是作为一种有效的近似方法,计算结果的相对值是非常准确的,不影响能带和态密度的分析。
对比图2(a)和(b)可知掺杂后导带和价带明显下移,其中导带下移 1.261 eV,价带下移 1.267 eV,导带下移程度略小于价带下移程度,从而使禁带变宽,但幅度不大。掺杂后TiO2导带宽度由1.324 eV减小至1.160 eV,导带的轨道增加,但能量范围减小;价带宽度由4.593 eV增加到4.853 eV,价带的轨道分裂更大,能级增加数明显增多,能量范围扩大。同时在禁带中引入一条明显的杂质能级,此杂质能级跨越费米能级,波动幅度为0.284 eV,如此小的波动使得从价带中跃迁到中间杂质能级的电子不容易返回到价带,可以作为价带电子跃迁至导带的桥梁,使得价带电子可以首先被低能量光子激发跃迁至杂质能级,继续吸收光子后跃迁至导带,降低了光子吸收能量,从而表现为对可见光的吸收;另一方面来说,禁带中出现的杂质能级可作为光生电子和空穴的捕获势阱,减小电子-空穴的复合几率,如Reddy等[21]认为 Bi掺入 TiO2引起 Bi3+δ+的生成, 而 Bi3+δ+引起杂质能级的产生,这将捕获电子,利于电子-空穴对的分离,电子空穴移动到表面参加光催化反应,因此可提高TiO2的光催化活性。
2.4 态密度
图3(a~c)是掺杂前后锐钛矿相TiO2的分态密度(PDOS)图。由于影响半导体物理性质的主要是费米能级附近的电子结构,所以本文电子态密度能量显示范围为从-7~5 eV。由图3(a~b)可知掺杂前后TiO2导带主要由Ti3d态电子组成,价带主要由O2p和Ti3d态电子共同组成。由图3(b)可知,Bi掺杂TiO2的禁带中出现一小态密度峰,此峰对应于能带结构图中的杂质能级,为了更好地认识Bi掺杂对禁带宽度的影响,图 3(c)绘出了Bi掺杂TiO2能量在-7~5 eV的精细分态密度分布图。由图3(c)可知,此杂质能级主要由Bi6s、O2p和Ti3d态电子杂化而成。除此之外Bi的6s轨道对导带和价带都有贡献,在-4.727、-2.195、-3.591 和 2.285 eV 处有 4 个明显的态密度峰,但相对于杂质能级处的峰值小很多,这些是由于Bi6s轨道参与杂化形成的;Bi的6p轨道在-5.928、-4.857、-3.234、-2.275、2.480 eV 处分裂,形成5个明显的态密度峰,其中-5.928 eV处的态密度峰达到最大值。态密度峰值越大,说明此位置处Bi6p轨道和O2p、Ti3d、Ti4s轨道杂化很明显。
2.5 光学性质
为了揭示掺杂后锐钛矿相TiO2电子结构与光学性质之间的关系,为进一步研究TiO2晶体的光吸收性能等光学性质提供基础,我们计算了非极性多晶态TiO2的介电函数和光吸收性质。
2.5.1 介电函数虚部
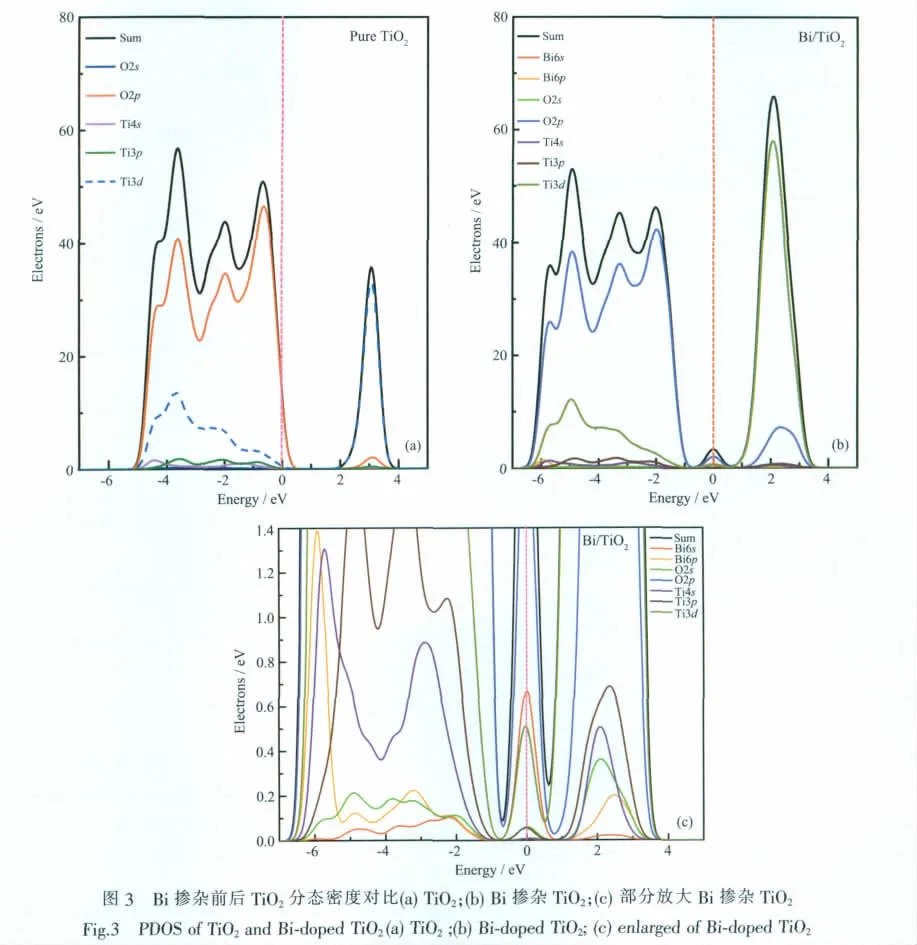
介电函数虚部ε2是能带体系最直接的表现形式,它反映了能级间电子跃迁所产生光谱的发光机理。Castep中介电函数虚部具体定义为:
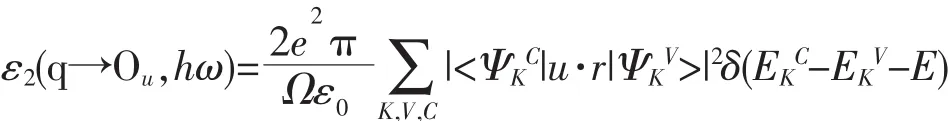
其中C、V分别表示导带和价带,K为倒格矢,u是入射电场的极化方向矢量,<ΨKC|u·r|ΨKV>为动量跃迁矩阵,EKC、EKV分别为导带和价带上的本征能级。由于电子结构中无论是带间跃迁还是带内跃迁的频率都远超过声子频率,在计算中仅考虑了电子跃迁,故介电函数可以表述为线性响应函数。从某种意义上说,介电响应函数比宏观光学常数更能表征材料的物理特性,更易于和物理过程的微观模型及固体的微观电子结构联系起来[20]。
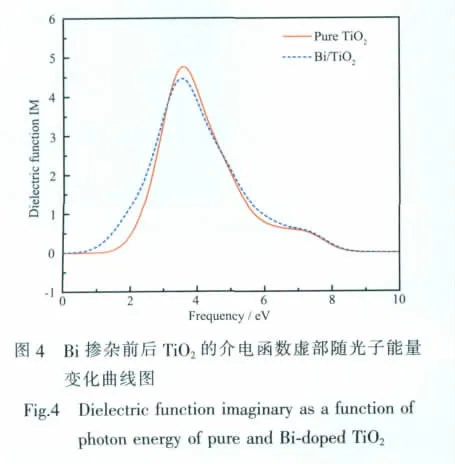
图4为纯锐钛矿相TiO2以及Bi掺杂TiO2的介电函数虚部随光子能量变化图。与未掺杂TiO2相比,Bi掺杂后谱线在低能方向出现明显红移,在高能方向发生少量蓝移。观察图4可发现,Bi掺杂TiO2的介电虚部谱接近于未掺杂TiO2,都存在一个由于电子跃迁引起的主介电峰,两峰相差0.025 eV(分别位于 3.520 7 eV 和 3.545 2 eV 处), 这是因为Bi掺杂后电子吸收光子能量而发生跃迁的过程中杂质能级起到桥梁作用,降低了跃迁所需能量,导致介电虚部峰左移。
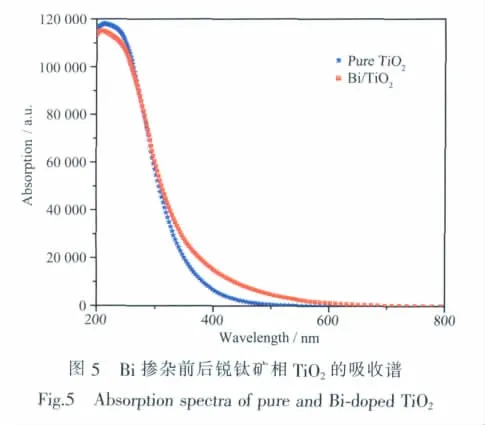
2.5.2 吸收光谱
图5是纯锐钛矿相TiO2和Bi掺杂TiO2的吸收光谱对比图,为了使计算结果跟实际相符,在计算光吸收性质时使用了剪刀差进行修正,修正值取1.072 eV(禁带宽度实验值与计算值之差)。由图可知在279 nm处出现交叉,Bi掺杂后TiO2吸收光谱相比于未掺杂TiO2的吸收光谱出现明显的吸收带边红移,同时在可见光区吸收强度增强,这与Huang[12]和Reddy等[21]在实验上观察到的吸收光谱红移现象一致,并且吸收光谱非常吻合。根据能带和态密度的结果,本文认为这是由于掺杂后禁带中出现由Bi引起的杂质能级充当了过渡能级,使得价带电子在光电子激发下可以首先跃迁至杂质能级,然后再跃迁至导带,降低了电子跃迁所需能量。Bi掺杂后TiO2禁带宽度虽有较小幅度增大,不利于电子跃迁,但是禁带中杂质能级的引入抵消了禁带宽度略微增大带来的不利影响,说明Bi掺杂锐钛矿相TiO2中杂质能级对光吸收增强起主要作用,这对Bi掺杂锐钛矿型TiO2光催化剂的制备有一定的理论参考价值。
3 结 论
采用基于密度泛函理论的平面波超软赝势方法研究了Bi掺杂前后锐钛矿相TiO2晶体结构、能带、电荷布居、态密度和光吸收性质。计算结果表明:Bi掺杂后O的电荷布居数增大,Ti的电荷布居数有所下降导致Bi掺杂后O原子周围的电负性增加,Ti原子周围的电正性有所减弱。在TiO2禁带中引入由Bi6s、O2p和Ti3d态杂化而成的杂质能级,同时禁带宽度略微变大,但是由于杂质能级抵消了禁带宽度变大带来的不利影响,从而使掺杂后TiO2吸收带边出现红移,在可见光区域吸收明显增强。
致谢:本文主要研究人员均为河北大学刘保亭教授课题组成员,全部计算均采用刘保亭教授所购买的Accelrys公司的Materials Studio软件中的量子力学模块Castep完成,感谢刘保亭教授在论文写作和机理探讨方面提供的帮助。
[1]CHEN Xiao-Yun(陈孝云),LU Dong-Fang(陆东芳),SUN Lan(孙岚),et al.Chinese J.Inorg.Chem.(Wuji Huaxue Xuebao),2012,28(2):307-313
[2]Lin Y M,Jiang Z Y,Zhu C Y,et al.Appl.Phys.Lett.,2012,101:062106
[3]Run L,Niall J.Chem.Mater.,2010,22(5):1616-1623
[4]Fu C,Li T Z,Qi J S,et al.Chem.Phys.Lett.,2010,494:117-122
[5]DAI Gao-Peng(戴高鹏),LIU Su-Qin(刘素芹),LUO Tian-Xiong(罗天雄),et al.Chinese J.Inorg.Chem.(Wuji Huaxue Xuebao),2012,28(8):1617-1622
[6]Guo M L,Du J L.Physica B,2012,407:1003-1007
[7]Kuwauchi Y,Yoshida H,Akita T,et al.Angew.Chem.Int.Ed.,2012,51(31):7729-7733
[8]Ding Y,Xu X J,Gan Z H,et al.Solid State Phenomena.,2011(181/182):348-351
[9]Gao Y L,Liu S Y,Zhang F,et al.Key Eng.Mater.,2012,509:65-73
[10]Deng Q R,Xia X H,Guo M L,et al.Mater.Lett.,2011,65(13):2051-2054
[11]Zhang Q Y,Li Y,Ackerman E A,et al.Appl.Catal.A:Gen.,2011,400(1-2):195-202
[12]Huang J H,Cheuk W,Wu Y,et al.J.Nanotechnol.,2012,2012:1-7
[13]Rengaraj S,Li X Z,Tanner P A,et al.J.Mol.Catal.A:Chem.,2006,247:36-43
[14]LIANG Xiao-Ming(梁晓明),ZHANG Su-Min(张苏敏),SHI Zai-Feng(史载锋).J.Hainan Normal Univ.(Hainan Shifan Daxue Xuebao),2011,24(4):422-424
[15]Xu X H,Wang M,Hou Y,et al.J.Mater.Sci.Lett.,2002,21:1655-1656
[16]Howard C J,Sabine T M,Dickson F.Acta Cryst.B:Struct.Sci.,1991,47:462-468
[17]YANGKe-Song(杨可松).Thesis for the Doctorate of Shandong University(山东大学博士论文).2010.
[18]Stampfl C,Van de Walle C G.Phys.Rev.B,1999,59(8):5521-5535
[19]Perdew J P,Mel L.Phys.Rev.Lett.,1983,51(20):1884-1887
[20]ZHAO Zong-Yan(赵宗彦),LIU Qing-Ju(柳清菊),ZHU Zhong-Qi(朱忠其),et al.Acta Phys.Sin.(Wuli Xuebao),2008,57(6):3760-3768
[21]Reddy P A K,Srinivas,Kala P,et al.Mater.Res.Bull.,2011,46:1766-1771
猜你喜欢
吉首大学学报(自然科学版)(2023年6期)2023-12-22 08:18:20
装备维修技术(2021年36期)2021-10-25 13:21:04
弹箭与制导学报(2021年3期)2021-07-30 02:56:52
矿产勘查(2020年8期)2020-12-25 02:46:36
原子与分子物理学报(2020年5期)2020-03-17 07:00:14
网印工业(2019年4期)2019-05-21 06:41:58
重型机械(2019年2期)2019-04-28 11:52:04
吉首大学学报(自然科学版)(2018年3期)2018-07-03 03:14:12
Chinese Journal of Chemical Engineering(2017年5期)2017-05-28 10:22:54
无机盐工业(2017年6期)2017-03-11 14:43:47
