
HPLC测定咳特灵胶囊中马来酸氯苯那敏的含量及均匀度
2013-09-27 00:50王娇杨宝翠姜国志陈钟
中国现代中药 2013年7期
王娇,杨宝翠,姜国志,陈钟
(神威药业集团有限公司,河北 石家庄 051430)
HPLC测定咳特灵胶囊中马来酸氯苯那敏的含量及均匀度
王娇*,杨宝翠,姜国志,陈钟
(神威药业集团有限公司,河北 石家庄 051430)
目的:建立咳特灵胶囊中马来酸氯苯那敏的高效液相色谱分析方法。方法:C18柱,乙腈-0.3%十二烷基硫酸钠溶液-磷酸(60∶40∶0.02)(用三乙胺调节pH值至3.3±0.1)为流动相,流速为1.0 mL·min-1;于225 nm测定咳特灵胶囊中的马来酸氯苯那敏含量。结果:马来酸氯苯那敏的线性范围为156.08~936.48 μg,平均回收率为98.23%(RSD=0.12%)。结论:该方法简便、准确、无干扰,可用于咳特灵胶囊中马来酸氯苯那敏的含量及含量均匀度的测定。
高效液相色谱法;咳特灵胶囊;马来酸氯苯那敏;含量均匀度
咳特灵胶囊收载于《部颁标准·中药成方制剂》第十四册,组方为小叶榕干浸膏与马来酸氯苯那敏,具有镇咳、祛痰、平喘、消炎的作用,用于治疗咳喘及慢性支气管炎咳嗽,疗效确切。该品种每粒含马来酸氯苯那敏仅1.4 mg,处方量较小,标准中只对马来酸氯苯那敏的限度进行检查,要求吸收值范围在0.35~0.65,范围较宽,并且该方法操作复杂,误差较大。《中国药典》2010年版二部[1]规定,硬胶囊剂每粒标示量不大于25 mg者,均应检查含量均匀度。为更好地控制产品质量,参考有关文献[2-8],建立了马来酸氯苯那敏的含量及含量均匀度检测方法,该方法简便、准确、无干扰,现介绍如下。
1 仪器与试药
Ultimate 3000高效液相色谱仪(美国戴安公司);CPA2250型分析天平(Sartorius公司)。马来酸氯苯那敏对照品(批号:100047-200606,含量99.7%,中国食品药品检定研究院);乙腈为色谱纯;其他试剂均为分析纯。咳特灵胶囊(神威药业集团有限公司,批号:1108021,1108022,1108023)。
2 方法与结果
2.1 色谱条件及系统适用性试验
色谱柱为Phenomenex C18(250 mm×4.6 mm,5 μm);流动相为乙腈-0.3%十二烷基硫酸钠溶液-磷酸(60∶40∶0.02)(用三乙胺调节pH值至3.3±0.1);流速为1.0 mL·min-1;进样量为20 μL;检测波长为225 nm。理论板数按氯苯那敏峰(色谱图中马来酸峰在前,氯苯那敏峰在后)计算不低于2 000。
2.2 对照品溶液的制备
精密称取105 ℃干燥3 h的马来酸氯苯那敏对照品适量,加乙腈溶解并定量稀释制成每1 mL中约含60 μg的溶液,精密量取5 mL,置10 mL量瓶中,加0.3%十二烷基硫酸钠溶液稀释至刻度,摇匀即得。
2.3 供试品溶液的配制
取本品内容物,研细,称取0.5 g,精密称定,置具塞锥形瓶中,精密加入乙腈25 mL,密塞,称定重量,超声处理1 h(功率300 W,频率40 kHz),放冷,再称定重量,用乙腈补足减失的重量,摇匀,滤过,精密量取续滤液5 mL,置10 mL量瓶中,加0.3%十二烷基硫酸钠溶液稀释至刻度,摇匀即得。
2.4 专属性试验
按处方组成、制法制成缺马来酸氯苯那敏的阴性样品,同供试品溶液制备方法制得阴性供试品溶液。结果阴性供试品溶液与对照品溶液、样品溶液色谱保留时间相应的位置上无其他色谱峰出现,说明其他成分和辅料对测定无干扰。见图1~3。

图1 马来酸氯苯那敏对照品HPLC图

图2 咳特灵胶囊样品HPLC图

图3 咳特灵胶囊阴性样品HPLC图
2.5 线性关系的考察
精密称取105 ℃干燥3 h的马来酸氯苯那敏对照品适量,加乙腈溶解并定量稀释制成每1 mL中约含60 μg的溶液,精密量取5 mL,置10 mL量瓶中,加0.3%十二烷基硫酸钠溶液稀释至刻度,摇匀,作为对照品溶液。精密量取5,10,15,20,25,30 μL注入液相色谱仪,按上述色谱条件测定。以对照品溶液的量为横坐标(X),峰面积为纵坐标(Y),绘制标准曲线,得回归方程为Y=0.032 4X-0.040 9,r=0.999 9。结果表明,马来酸氯苯那敏在156.08~936.48 μg线性关系良好。
2.6 重复性考察
取对照品溶液,精密量取20 μL注入液相色谱仪,连续测定6次,结果马来酸氯苯那敏峰面积RSD=0.081%,说明含量测定方法的重复性良好。
2.7 溶液稳定性考察
取供试品溶液,分别于0,2,4,8,16 h精密量取20 μL注入液相色谱仪,结果马来酸氯苯那敏峰面积RSD=1.10%,说明供试品溶液在16 h内稳定。
2.8 重现性考察
制备供试品溶液6份,分别精密量取20 μL注入液相色谱仪,测得峰面积并计算含量,结果马来酸氯苯那敏的平均含量为1.374 mg/粒,RSD=1.98%。
2.9 回收率试验
精密称取处方量80%、100%、120%的马来酸氯苯那敏,制成模拟样品,精密称取不同水平模拟样品各3份,测定含量,并计算回收率,结果见表1。
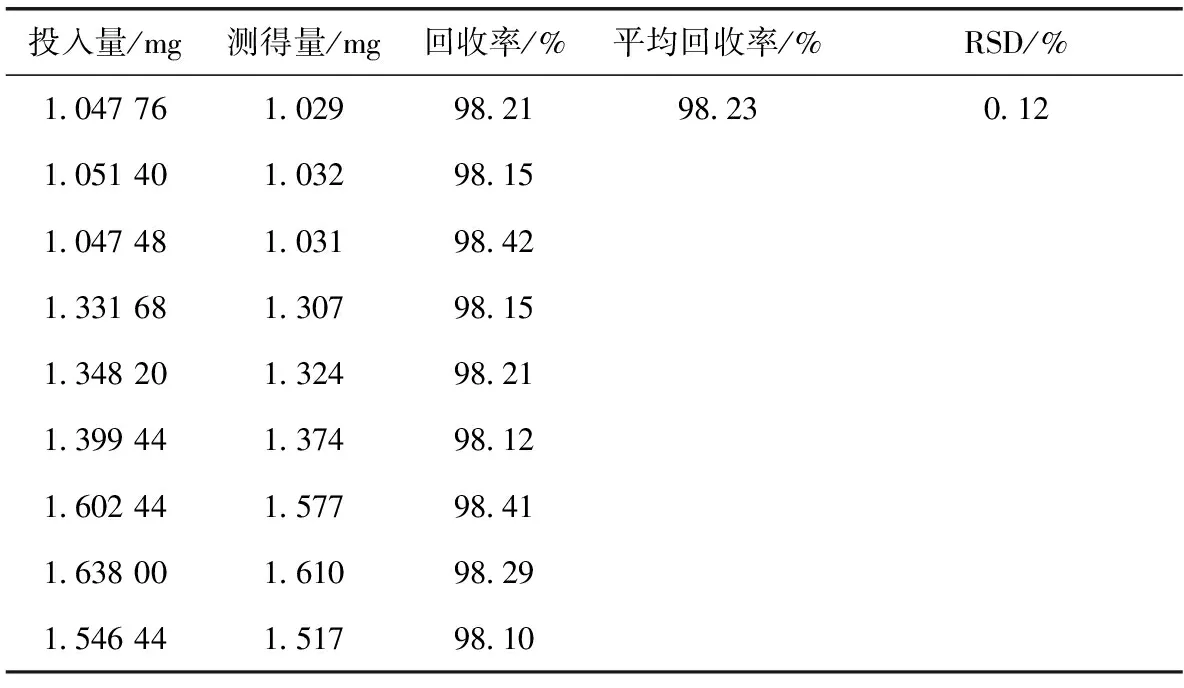
表1 马来酸氯苯那敏回收率试验
2.10 样品含量测定
将1108021,1108022,1108023 3批样品按2.3方法分别制成供试品溶液,各精密量取20 μL注入液相色谱仪,测得峰面积并计算含量,结果分别为1.284,1.281,1.275 mg/粒。
2.11 样品含量均匀度测定[1]
取供试品1粒,依2.3供试品溶液的制备方法从“内容物置具塞锥形瓶”开始操作。1108021,1108022,1108023 3批样品分别制成10个供试品溶液,各精密量取20 μL注入液相色谱仪,测得峰面积并计算含量均匀度,A+1.8S分别为7.2,7.0,6.6,均小于15.0,说明含量均匀度符合规定。
3 结论与讨论
实验中,比较了提取时间对提取效果的影响,确定超声1 h提取最完全。由二极管阵列检测器获得的氯苯那敏色谱峰UV扫描图谱得知,在225 nm处有最大吸收,因此选择检测波长为225 nm。
参考大量文献,池浩波等[3]处理样品时用水溶解,调pH值,用三氯甲烷、正己烷提取,饱和氯化钠水洗等;简淑娟[4]处理样品时三氯甲烷超声,水浴蒸干,甲醇定容;范蕾等[5]处理时二氯甲烷冷浸,蒸干,稀盐酸定容;周娟等[6]处理样品时无水乙醇超声,反复洗涤、滤过、超声等,这些处理方法复杂,操作误差大。本文建立的处理方法只需乙腈超声处理,0.3%十二烷基硫酸钠溶液定容,操作简便。
本文建立的方法简便、准确、无干扰,可用于测定咳特灵胶囊中马来酸氯苯那敏的含量及含量均匀度。
[1] 国家药典委员会.中国药典[S].二部.北京:中国医药科技出版社,2010:附录88.
[2] 国家食品药品监督管理局.国家药品标准·酚氨咖敏颗粒[S].WS-10001-(HD-1108)-2002.
[3] 池浩波,曾伟杰.HPLC法测定咳特灵胶囊(片)中马来酸氯苯那敏的含量[J].广东药学,2003,13(4):19-21.
[4] 简淑娟.HPLC法测定咳特灵片及胶囊中氯苯那敏的含量及含量均匀度[J].中国药师,2005,8(4):313-314.
[5] 范蕾,赵娟,王兰,等.HPLC法测定咳特灵胶囊中马来酸氯苯那敏的含量[J].中医药导报,2006,12(11):62-63.
[6] 周娟,朱玲.HPLC法测定咳特灵胶囊中马来酸氯苯那敏的含量及含量均匀度[J].安徽医药,2010,14(4):406-407.
[7] 江溱. HPLC法测定咳特灵糖浆中马来酸氯苯那敏的含量及含量均匀度[J].中国民族民间医药,2011,(18):50-51.
ContentUniformityDeterminationofChlorphenamineMaleateinKetelingCapsulesbyHPLC
WANG Jiao,YANG Bao-cui,JIANG Guo-zhi,CHEN Zhong
(ShinewayPharmaceuticalGroupLtd.,Shijiazhuang051430,China)
Objective: To establish HPLC method for content uniformity determination of chlorphenamine maleate in Keteling Capsules.Methods: C18column was used,the mobile phase is acetonitrile-0.3% SDS-phosphoric acid (60∶40∶0.02,adapt pH to 3.3 with triethylamine),the flow rate is 1.0 mL·min-1,detecting wavelength is 225 nm.Results: The linear range of chlorphenamine maleate was 156.08~936.48 μg. The average recovery was 98.23% with RSD 0.12%.Conclusion: The method is simple,sensitive,rapid and accurate,can be used for the quality control of Keteling Capsules.
HPLC; Keteling Capsules; Chlorphenamine maleate; Content uniformity
2013-02-26)
*
王娇,E-mail:wangjiaowangjiao1212@126.com
猜你喜欢
四川大学学报(自然科学版)(2022年1期)2022-02-10
环境卫生工程(2021年1期)2021-03-19
中国医药指南(2020年31期)2020-12-30
河南畜牧兽医(2020年21期)2020-01-10
中国农村水利水电(2018年3期)2018-04-13
北京航空航天大学学报(2017年3期)2017-11-23
中国药物滥用防治杂志(2017年3期)2017-01-13
纺织检测与标准(2016年3期)2016-12-16
中国医疗美容(2015年5期)2015-02-03
