
密度泛函理论研究十二烷硫醇在Au(111)面上的吸附
2013-09-21 09:00:18范晓丽冉润欣杨永良
物理化学学报 2013年9期
范晓丽 冉润欣 张 超 杨永良
(西北工业大学,凝固技术国家重点实验室,西安710072)
1 引言
自组装单分子层膜(简称SAMs)是有机分子在固体表面自发形成的有机分子薄层.其中,有机分子在Au表面形成的稳定且有序结构在界面润湿、表面化学,以及生物化学传感器、隔膜等电子学领域有着广泛的应用.1−5因此,关于有机分子在Au表面的吸附成为表面科学和多相催化化学领域十分重要的研究内容.近年来,有机小分子在Cu,Ag,Au表面吸附的理论研究已经较为详尽.6−11其中,硫醇及二硫化物在Au基底上的吸附是最具有代表性,同时也是被研究得最多的体系.
最简单的烷基硫醇分子,甲烷基硫醇(CH3S)在Au(111)表面上的吸附吸引了绝大部分的关注.12−17目前的实验和理论研究一致表明,12−15甲烷硫醇分子(CH3SH)或二甲基二硫分子(CH3S―SCH3)在Au(111)表面沉积均可生成甲烷基硫醇SAM.两个过程均由未解离的分子吸附开始,当基底表面温度升高时,S―H键或S―S键解离,生成稳定吸附的CH3S基团,CH3S基团是甲烷基硫醇SAM的基本组成单元.在最初的分子吸附状态,甲烷硫醇分子吸附在Au(111)表面的top位上,吸附能为0.34 eV.当S―H键解离后,CH3S基团吸附在bridge位偏向fcc空位的地方,吸附能为2.03 eV.13,16
相较于甲烷硫醇SAMs,对含碳原子数超过六的长链硫醇分子的研究则比较少.18−20Yang课题组18采用团簇模型模拟了含碳原子数从六到十一的烷烃硫醇在Au(111)表面中空位吸附的扫描隧道显微镜(STM)图像,尽管来源于分子和基底相互作用的新电子态出现在费米能级附近,但模拟结果表明STM图像主要显示吸附分子末端的电子结构.Wang和Selloni19计算了n=1−8(碳原子数)烷烃硫醇在Au(111)表面不同位置吸附的结构和能量.最近,Nakayz等20采用STM实验技术研究了Au(111)表面八碳硫醇分子SAMs的表面结构,发现退火温度为70、120和150°C时的表面结构分别为倾斜角为~30°的直立结构、平铺结构和“lattice-gas”相.这些研究工作均集中在H解离后的表面吸附现象,并没有涉及分子最初到达表面时的吸附情况和表面结构.
本文以十二烷硫醇分子为对象,采用密度泛函理论和周期性平板模型方法,通过计算该分子在Au(111)面上不同位置以不同角度吸附的各种可能构型,系统地研究S―H键未解离时C12H25SH分子和解离后的C12H25S基团在Au(111)面上的吸附,讨论长链十二烷硫醇在Au(111)表面的优先吸附位置和构型,对比解离前和解离后吸附体系的总能量,分析S―H键断裂的可能.该研究充分考虑了吸附位置和空间构象等因素对解离和未解离吸附的影响,并且通过电子结构计算判断S―H键解离前后的吸附性质,同时与短链硫醇在Au(111)表面上的吸附做比较,讨论硫醇分子链长度对其吸附位置以及吸附能的影响.
2 计算方法
本文所有的计算均采用基于密度泛函理论(DFT)21,22的从头算量子力学程序包VASP(Vienna ab initio simulation Package)23−26进行.计算时采用平面缀加波PAW(Projector augmented wave)赝势27,28描述离子实与价电子之间的相互作用,电子交换关联能采用广义梯度近似(GGA)的PBE29方法.计算中选用Monkhorst Pack网格,经过测试将k网格大小设为3×3×1,截断能设为400 eV.结构优化过程中的收敛条件为原子所受的最大作用力小于0.3 eV·nm−1.
本文采用平板模型来模拟Au(111)表面.计算中所取的超单胞大小为4×4,边长为1.1728 nm×1.1728 nm,平板模型的厚度为六个原子层,z方向真空区厚度为2.5 nm.本文计算单个十二烷硫醇分子在(4×4)的Au(111)表面的吸附,对应的覆盖度为1/16,因此吸附分子之间的van der Waals相互作用可以忽略不计.在所有的计算中,十二烷硫醇分子和表面的5层Au原子完全松弛,最底层Au原子则固定在体相位置上.
我们计算十二烷硫醇分子的部分结构参数为:S―C键的键长为0.168 nm,S―H键的键长为0.157 nm,C―S键与相邻的S―H键之间的夹角为94.69°,C―C键的键长为0.153 nm左右,C―H键键长为0.110 nm左右,C―H键之间的夹角为105.9°左右.
3 结果与讨论
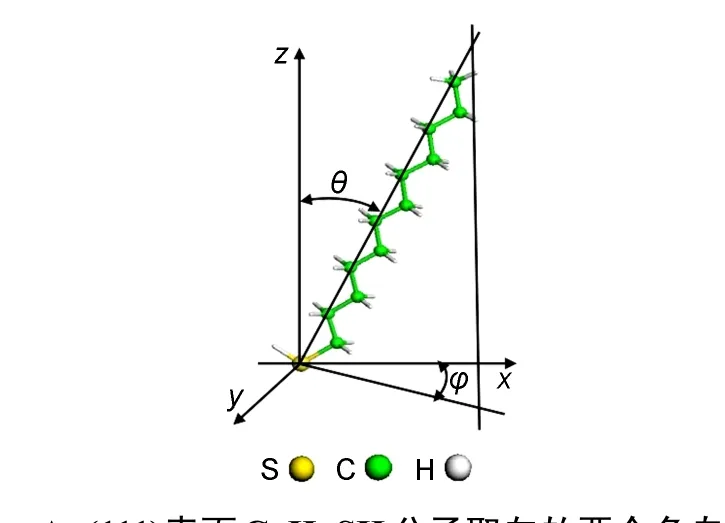
图1 Au(111)表面C12H25SH分子取向的两个角自由度Fig.1 Angle degrees of freedom of C12H25SH onAu(111)surface
如图1显示,硫醇分子在Au(111)表面的吸附结构主要由三个参数决定:末端S原子在表面的吸附位置;分子链的倾斜角θ,即分子链与表面法线方向的夹角;分子链的倾斜方向φ,即分子链在表面的投影方向.Au(111)表面有三种对称的吸附位置:top位;fcc和hcp空穴位;bridge位(简称bri).如图2所示,top位为表面Au原子正上方的位置;bri位为最近邻的两个金原子的中点位置;fcc空穴位是从表面看进去第三层原子的正上方位置;hcp空穴位则为第二层原子的正上方位置.除了以上三种对称位置以外,还有一些可能的吸附位置,比如:bri-hcp,brifcc分别为bri偏向hcp或fcc的位置,而top-bri位则是top位偏向bri的位置.
3.1 C12H25SH分子在Au(111)表面的未解离吸附
根据以往的计算结果,30,31硫醇分子的倾斜方向对吸附结构和能量的影响较小,因此本文忽略这个因素,只考虑了决定硫醇分子吸附结构的前两个因素,即吸附位置和倾斜角度.我们分别以四个倾斜角度0°、30°、60°和90°,将C12H25SH分子置于四个可能的吸附位置(top、hcp、fcc、bri)上,构建了16个初始构型.
对于这16个初始构型,我们通过能量最小化来优化吸附结构,计算结果列于表1.其中,C12H25SH分子在Au(111)表面未解离吸附的吸附能是根据吸附前后体系总能量的变化进行的,所用的公式如下:
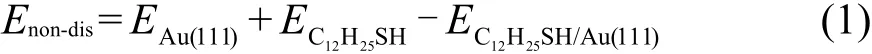

图2 Au(111)表面典型吸附位置Fig.2 Typical sites onAu(111)surface
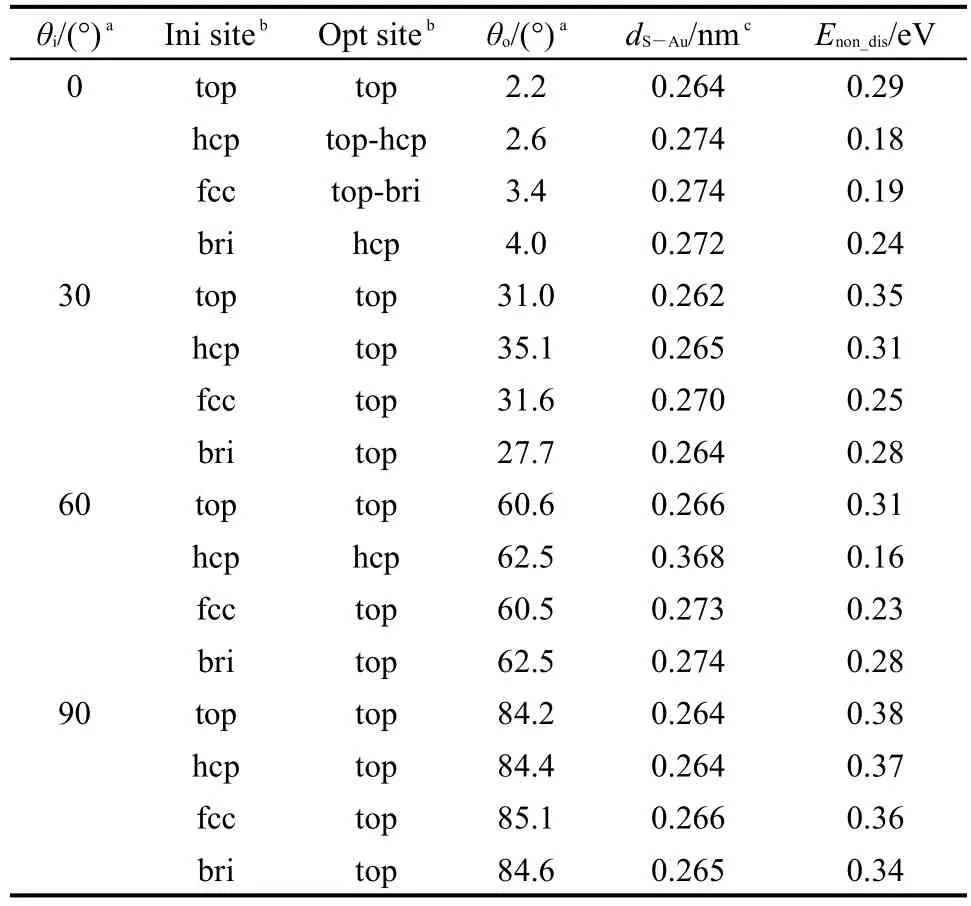
表1 C12H25SH在Au(111)表面未解离吸附构型的结构参数和吸附能量计算结果Table 1 Computational results on the atomic structures and energies for the non-dissociative adsorptions of C12H25SH onAu(111)surface
其中,EAu(111)和EC12H25SH分别表示Au(111)基底和单个C12H25SH分子的能量,而EC12H25SH/Au(111)则表示吸附体系的总能量.根据公式的定义,正的Enon-dis值表示放热反应,负的Enon-dis值则表示吸热反应.
表1中的数据显示C12H25SH分子在Au(111)表面的吸附为放热反应.对比四个不同位置上吸附的能量,我们发现C12H25SH分子倾向于吸附在top位,吸附能量为0.23−0.38 eV,与Au原子之间的距离为0.262−0.273 nm.在所有的稳定吸附构型中,除了倾斜角在30°−60°之间的直立构型外,我们还发现了一种平铺构型,该类构型中分子的倾斜角度为84°−85°,吸附能量为0.34−0.38 eV.
图3(a)显示了最稳定的直立吸附构型(30-top),该构型的倾斜角度为31°,吸附能量为0.35 eV,S原子与表面被吸附Au原子之间的距离为0.262 nm.图3(b)则显示了最稳定的平铺吸附构型(90-top),该构型的倾斜角度为84°,吸附能量为0.38 eV,S原子与表面被吸附Au原子之间的距离为0.264 nm.直立吸附构型中,分子链的结构较吸附前几乎没有变化,但是平铺构型中,分子链稍有弯曲.平铺构型的吸附能量比直立构型的吸附能量大0.03 eV,我们判断该微小差别来自于平铺构型中碳链上的部分H原子与表面之间的相互作用.图3(b)中给出了部分H原子与表面Au原子之间的距离.

图3 C12H25SH分子在Au(111)表面的未解离吸附构型Fig.3 Non-dissociative adsorption geometries of C12H25SH onAu(111)surface
表1的数据显示60-hcp构型的吸附能量为0.16 eV,S原子与表面最近邻原子之间的距离为0.368 nm.该构型中S―Au原子之间的距离大于S原子的范德华半径(0.180 nm)和Au原子的范德华半径(0.166 nm)之和,结合吸附能量的数值,我们判断该吸附构型属于物理吸附.此外,S原子半径为0.10 nm,Au原子的半径为0.179 nm,S原子在top位吸附的构型中S原子与被吸附的Au原子之间的距离稍小于S、Au两原子半径之和,应该属于弱的化学吸附.在下文中我们将会进一步通过电子结构计算来分析这些未解离吸附构型的性质.
以往的计算和实验研究结果显示,S―H键未断裂时,甲烷硫醇13,16,32和苯硫醇33,34分子均吸附在Au(111)表面的top位,与本文对于十二烷硫醇分子的计算结果一致.甲烷硫醇分子在top位的吸附能量为0.34−0.38 eV,16,32S原子与表面被吸附Au原子之间的距离为0.276 nm,十二烷硫醇分子在top位的吸附能量为0.38 eV,S原子与表面被吸附Au原子之间的距离为0.264 nm.这些结果表明,硫醇分子碳链长度对未解离吸附能量的影响不大,但是随着碳链长度的增加,S原子与表面Au原子之间的距离稍有减小.
3.2 C12H25S基团在Au(111)表面的解离吸附
C12H25SH分子吸附在Au(111)表面后,S原子与表面Au原子之间的相互作用使得S―H键减弱.实验证明STM电流脉冲和表面温度提升均可以促使S―H键断裂.13计算研究35表明硫醇分子RSH在Au表面均裂,即解离成RS自由基和H原子.S―H键解离后,H原子要么以H2分子的形式从表面解离,要么吸附在Au(111)表面.本文该部分计算H原子解离后,C12H25S基团在Au(111)表面的吸附.由于C12H25S是自由基,计算中考虑了自旋极化.
我们采用了与计算未解离吸附类似的方法,以四个倾斜角度0°、30°、60°和90°,将C12H25S基团分别置于四个可能的吸附位置上,构建了16个初始吸附构型.这16个构型的计算结果列于表2.C12H25S基团在Au(111)表面吸附能的计算采用如下公式:


表2 解离后的C12H25S吸附在Au(111)表面的结构参数和吸附能量计算结果Table 2 Computational results on the atomic structures and energies for the dissociated adsorption of C12H25S onAu(111)surface
其中,Edis表示C12H25S基团在Au(111)表面的吸附能,EAu(111)表示Au(111)基底的能量,EC12H25S表示单个C12H25S基团的能量,而EC12H25S/Au(111)则是吸附体系的总能量.
从表2中的数据可以看出,S―H键解离后,C12H25S倾向于吸附在bri偏向中空位上.最稳定的吸附结构中,硫原子吸附在bri-fcc位,吸附能为2.05−2.09 eV,S原子和表面最近邻Au原子之间的平均距离约为0.246 nm,分子倾斜角为27°−60°.硫原子在bri-hcp位的吸附能量比bri-fcc位的吸附能量约小0.09 eV.表2中,我们同样发现了平铺吸附结构,该类构型中C12H25S分子链与表面法线方向的夹角约为82°,吸附能量为2.0 eV,比直立吸附构型的能量小0.09 eV.
图4(a)显示C12H25S在bri-fcc位的直立吸附构型(30-top),该构型的倾斜角度为26.9°,吸附能量为2.09 eV,S原子与表面最近邻Au原子之间的平均距离为0.246 nm.图4(b)则显示了C12H25S基团在brifcc位的平铺吸附构型(90-top),该构型的倾斜角度为80°,吸附能量为2.01 eV,S原子与表面最近邻Au原子之间的平均距离为0.247 nm.与未解离C12H25SH分子的吸附相似,直立吸附构型中,分子链的结构较吸附前几乎没有变化,但是平铺构型中,分子链稍有弯曲.
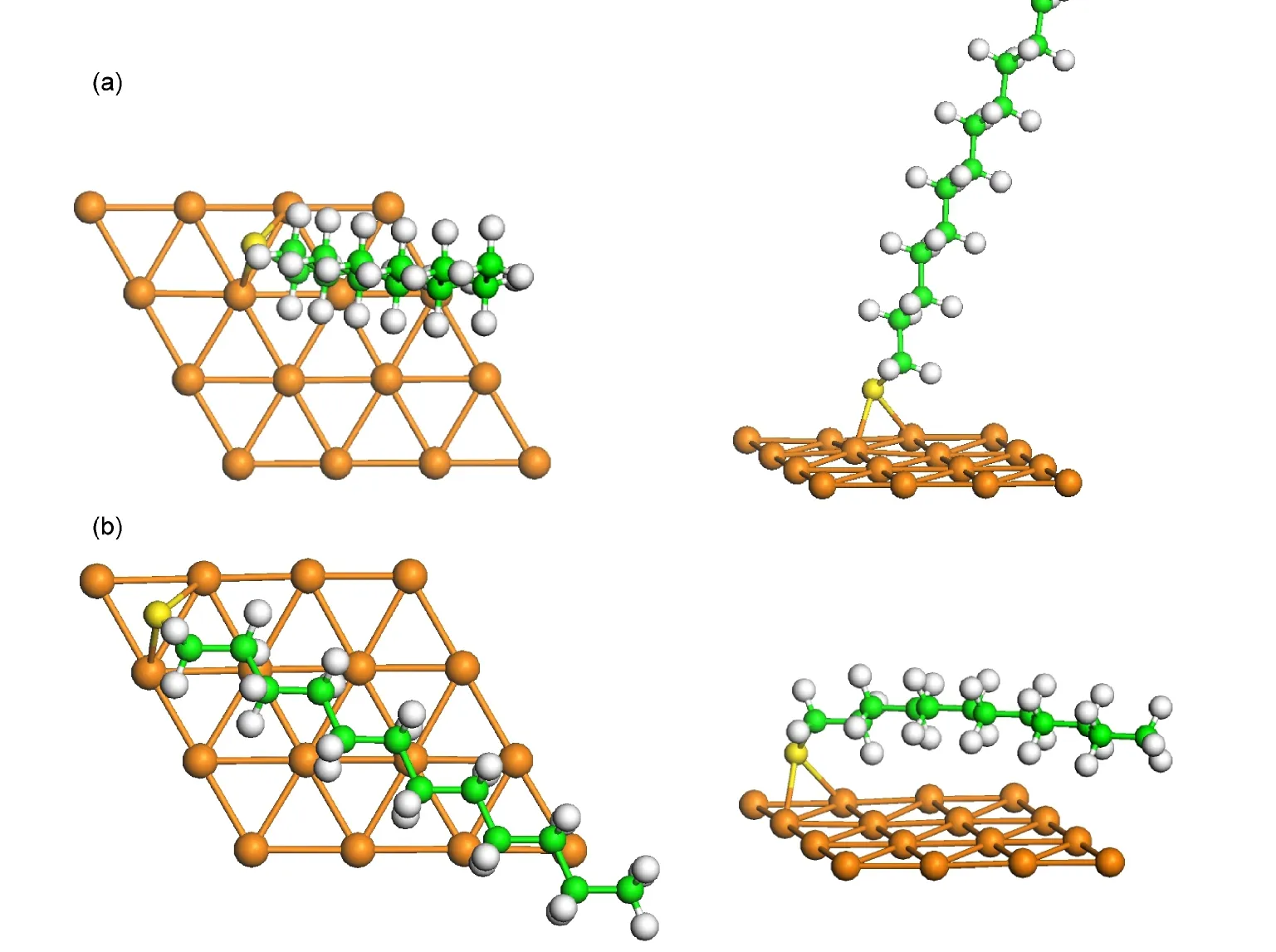
图4 C12H25S基团在Au(111)表面的吸附构型Fig.4 Adsorption geometries of C12H25S onAu(111)surface
本文发现的平铺构型是首次计算报道,以往的计算结果显示硫醇在Au(111)表面的倾斜角为30°−60°.对于C12H25S基团,直立吸附构型比平铺吸附构型稳定0.08 eV,因此直立构型为稳定存在的表面结构.我们的计算结果与最近的实验研究结果20一致,Nakayz等人发现辛烷硫醇在低温Au(111)的构型为倾斜角为30°左右的直立构型,当温度升高至150°C时,表面出现平铺构型,他们分析平铺构型是由部分硫醇的解离导致的.
根据以往的计算结果,甲硫醇吸附在Au(111)面的bri-fcc位,吸附能量为1.7−1.9 eV,36,37S原子与表面最近邻Au原子之间的距离(dS―Au)为0.252 nm;32丙硫醇在bri-fcc位的吸附能量为1.83 eV;36同时,Wang和Selloni19的计算表明含碳原子数为1−6的烷烃硫醇均吸附在Au(111)表面的bri-fcc位,吸附能量约为2.1 eV,dS―Au为0.247 nm.本文的计算结果显示,碳原子数为12的烷烃硫醇基团在Au(111)表面bri-fcc位的吸附能量为2.09 eV,dS―Au为0.246 nm,相比于短链硫醇,吸附能量略有增加,S原子与表面最近邻Au原子之间的距离稍有减小.
3.3 解离或未解离吸附
以上两个部分的计算显示C12H25S基团与Au(111)表面的吸附能比C12H25SH分子的吸附能大1.7 eV.表明C12H25S基团与Au(111)表面的相互作用远远强于C12H25SH分子与Au表面的相互作用.但是仅凭这些数据,我们还不能断定S―H键的解离.我们将计算比较两种解离吸附:S―H键断裂后,C12H25S基团和H原子均吸附在Au(111)表面,即C12H25S_H/Au(111);解离后的H原子以H2分子形式解离,即C12H25S/Au(111)+1/2H2.并与未解离吸附C12H25SH/Au(111)作比较,判断S―H键的解离以及H原子的去处.
对于第一种情况,我们首先计算了单个H原子在Au(111)表面的吸附,发现H原子在fcc空位上的吸附最稳定,吸附能为3.41 eV.随后,我们从表2中C12H25S基团的吸附结构中,选择四个稳定结构0-fcc、30-top、60-hcp、90-top,分别将H原子置于S―H键方向上的fcc位置,以此构建了4个可能的解离吸附C12H25S_H/Au(111)构型.计算结果列于表3.其中,dS―Au和dAu―H表示S原子和H原子与Au(111)表面最近邻Au原子之间的距离,EIdis表示C12H25S_H/Au(111)体系的解离吸附时的能量,计算公式如下:

表3 C12H25S_H在Au(111)表面吸附的结构参数和能量计算结果Table 3 Computational results on the atomic structures and energy for the dissociative adsorption of C12H25S_H onAu(111)surface

从表3的数据可知,解离吸附体系C12H25S_H/Au(111)的吸附能量均为负值,表示S―H键解离后H原子吸附在表面的反应为吸热反应.相较于C12H25S基团的吸附,我们发现H原子在附近的吸附并没有影响C12H25S基团的吸附位置和S原子与表面Au原子之间的距离.图5显示了C12H25S_H/Au(111)解离吸附的最稳定构型,相应的吸附能为−0.01 eV.该构型中,S原子吸附在bri-fcc位,S―Au距离为0.247 nm,H原子吸附在fcc位,H―Au距离为0.19 nm.
对于第二种解离吸附,我们选取最稳定的C12H25S/Au(111)构型,以此计算H原子以H2分子形式解离的吸附能量
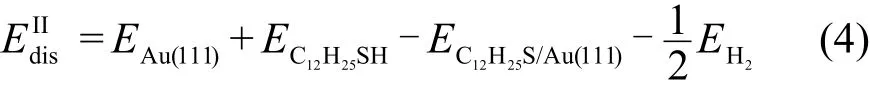
对比S―H键未断裂时C12H25SH/Au(111)的吸附能量(0.38 eV),和S―H键断裂后C12H25S_H/Au(111)的吸附能量(−0.01 eV),以及C12H25S/Au(111)+H2的吸附能量(0.19 eV),不难发现S―H键未断裂时的吸附相对于断裂后的吸附要稳定.据此我们判断低温时十二烷硫醇分子倾向于未解离吸附在Au(111)表面top位,但是当表面温度升高或者当STM针尖施加电流脉冲时,S―H键可能被解离.解离后的H原子以H2分子的形式离开表面.H原子解离后,C12H25S基团直立吸附在Au(111)表面,但是当表面温度继续升高时,可能出现平铺吸附构型.
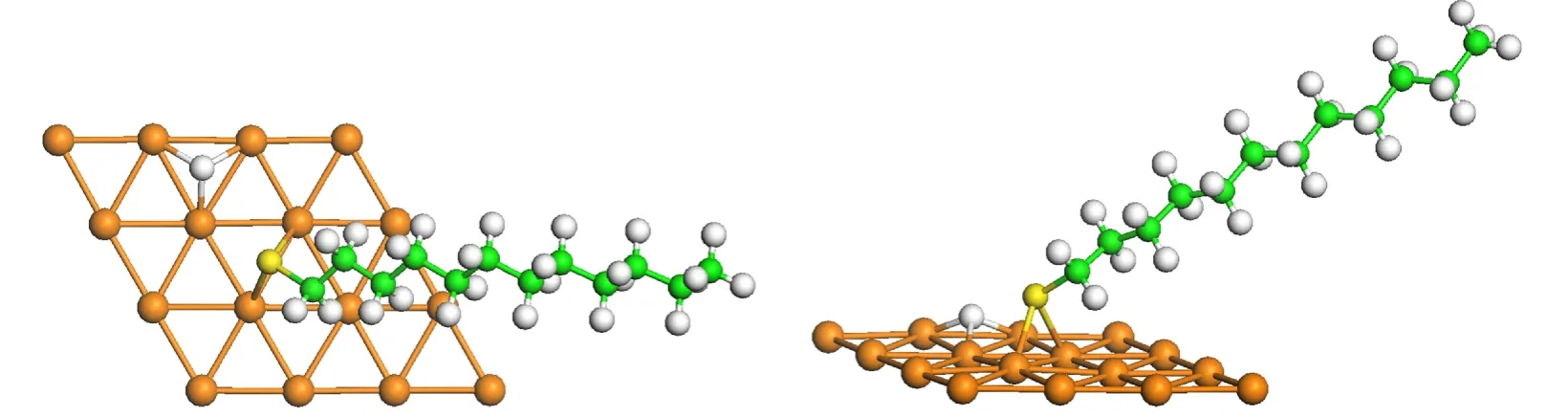
图5 C12H25S_H在Au(111)表面的吸附构型的顶视图和侧视图Fig.5 Top and side views of the dissociative adsorption geometry of C12H25S_H onAu(111)surface
3.4 电子结构分析
吸附结构和吸附能的计算结果表明C12H25SH分子在中空位吸附的60-hcp结构属于物理吸附,在top位的吸附属于弱化学吸附,S―H键断裂后C12H25S基团的吸附为化学吸附.本文该部分首先计算了这三种吸附结构的局域电子态密度(LDOS),通过分析电子结构的变化来讨论S原子和Au原子之间的相互作用.
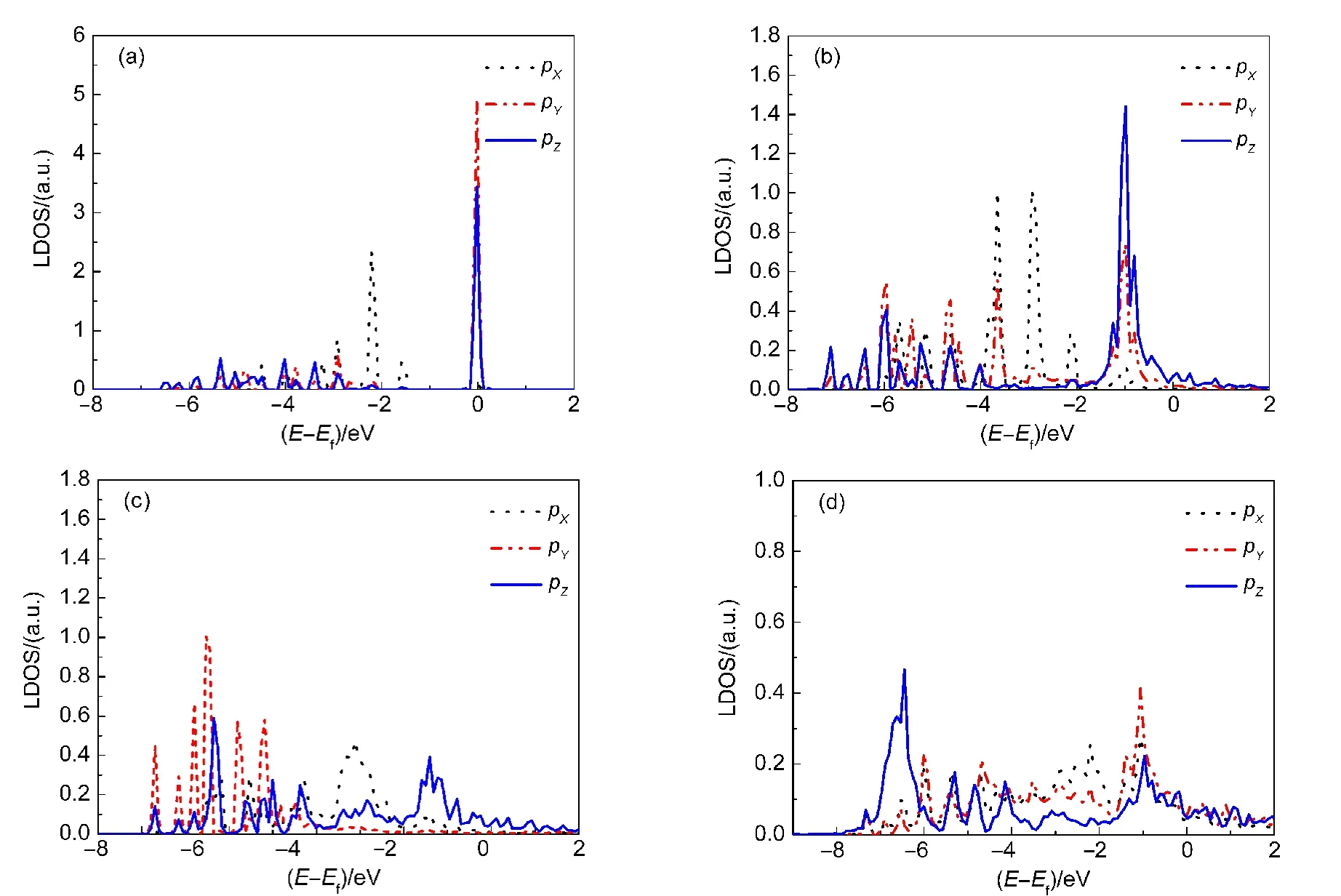
图6 S原子3p电子态在四种体系中的局域态密度(LDOS)图Fig.6 Decomposed local density of states(LDOS)on the 3p orbit of the S atom
图6所示为S原子3p电子态在单独的C12H25SH分子及吸附状态下的LDOS.如图6(a)所示,单独的C12H25SH分子中S原子的LDOS有多个规整的峰,对应于S原子的各个电子轨道,其中大多数的电子态都处在E−Ef=0 eV的位置上,表明S原子有很强的活性.图6(b)为C12H25SH分子在hcp中空位吸附的60-hcp构型的LDOS,相比于图6(a),该图中各个峰的位置均向低能量方向稍有移动,且峰的宽度稍有增加,但基本上还是规整的峰,表明C12H25SH分子在hcp中空位吸附的构型属于物理吸附.C12H25SH分子在top位吸附的30-top构型的LDOS被示于图6(c),我们发现E−Ef=0 eV处电子密度大幅度降低,且该位置附近出现新的能带群,此外,低能量能带群的密度高于高能量能带群的密度,这些现象表明C12H25SH分子在top位吸附的构型中,S原子与表面之间形成了新的成键轨道.图6(d)所示为C12H25S基团在表面吸附的30-top构型的LDOS,相较于图6(c),E−Ef=0 eV处电子密度进一步降低,而且新的能带群更加密集,这些变化说明S―H键断裂后S原子与表面之间成键的数目增加,而且键合更强.
吸附结构和能量以及电子结构的计算一致表明,C12H25SH分子吸附在Au(111)表面top位的稳定构型属于弱化学吸附;C12H25S基团在表面bri-fcc位吸附的构型属于化学吸附.虽然解离吸附C12H25S_H/Au(111)的稳定性相较于分子吸附C12H25SH/Au(111)的稳定性较差,但是S―H键断裂后,C12H25S基团与Au表面的相互作用更强,而且C12H25S/Au(111)构型是形成硫醇SAM的基本单元.
在电子态密度计算的基础上,我们进一步计算了C12H25SH分子以及C12H25S基团在Au(111)表面吸附的差分电荷密度图,结果如图7所示.关于差分电荷密度的定义如下:

其中,ρAB表示C12H25SH/Au(111)或C12H25S/Au(111)吸附总体系的电荷密度,ρA表示C12H25SH分子或C12H25S基团的电荷密度,ρB表示Au(111)表面即吸附基底的电荷密度.

图7 (a)C12H25SH分子和(b)C12H25S基团在Au(111)表面解离吸附的30-top构型的差分电荷密度图Fig.7 Charge density difference of(a)C12H25SH molecule and(b)C12H25S group adsorbed onAu(111)surface(30-top adsorption configuration)
图7 (a)和图7(b)相比可以看出,C12H25SH分子与Au表面之间的成键电荷明显比C12H25S基团与Au表面之间的成键电荷少,Bader analysis算法38得出图7(a)中电子转移0.04e,图7(b)中电子转移0.16e,表明H原子解离后,硫醇基团与表面的相互作用较解离前更强.
4 结论
通过计算S原子在Au(111)表面上不同位置以不同倾斜角度吸附的系列构型,研究了S―H键未解离时C12H25SH分子以及H原子解离后C12H25S基团在Au表面的吸附.对比两种解离吸附,我们发现C12H25SH分子和C12H25S基团均存在两种表面吸附构型,直立构型和平铺构型.未解离的C12H25SH分子倾向于吸附在top位,平铺构型的吸附能量为0.38 eV,比直立构型的吸附能量大0.03 eV,属于弱化学吸附;S―H键断裂后,C12H25S基团吸附在表面bridge偏向fcc的位置上,直立构型的吸附能量为2.09 eV,比平铺构型的吸附能量高0.08 eV.两种解离吸附稳定性均比未解离吸附的稳定性差.虽然如此,解离后C12H25S基团与Au表面的相互作用更强.
与短链(n=1−6)烷烃硫醇在Au(111)表面的吸附做对比,我们发现未解离的硫醇分子RSH(R表示烷基链)倾向于吸附在top位;S―H键解离后,RS基团倾向于吸附在表面的bri-fcc位.随着碳链长度的增加,吸附能量稍有增加,S―Au距离稍有减小.更重要的是,对于长链硫醇,未解离吸附直立构型和平铺构型的能量相差不大,两种构型可能共同存在;但是解离吸附的表面结构以直立构型为主,当温度升高导致部分硫醇从表面解离后,平铺吸附构型可能出现.
(1) Ulman,A.Chem.Rev.1996,96,1533.doi:10.1021/cr9502357
(2) Schreiber,F.Prog.Surf.Sci.2000,65,151.doi:10.1016/S0079-6816(00)00024-1
(3) Schreiber,F.J.Phys.:Condes.Matter 2004,16,R881.
(4) Love,J.C.;Estroff,L.A.;Kriebel,J.K.;Nuzzo,R.G.;Whitesides,G.M.Chem.Rev.2005,105,1103.doi:10.1021/cr0300789
(5) Nenchev,G.;Diaconescu,B.;Hagelberg,F.;Pohl,K.Phys.Rev.B 2009,80,081401.doi:10.1103/PhysRevB.80.081401
(6) Chen,W.K.;Cao,M.J.;Liu,S.H.;Xu,Y.;Li,Y.;Li,J.Q.Acta Phys.-Chim.Sin.2005,21,903.[陈文凯,曹梅娟,刘书红,许 莹,李 奕,李俊篯.物理化学学报,2005,21,903.]doi:10.3866/PKU.WHXB20050816
(7) Cao,M.J.;Chen,W.K.;Liu,S.H.;Lu,C.H.;Xu,Y.;Li,J.Q.Chin.J.Catal.2006,27,223.[曹梅娟,陈文凯,刘书红,陆春海,许 莹,李俊篯.催化学报,2006,27,223.]
(8) Cao,M.J.;Chen,W.K.;Liu,S.H.;Xu,Y.;Li,J.Q.Acta Phys.-Chim.Sin.2006,22,11.[曹梅娟,陈文凯,刘书红,许 莹,李俊篯.物理化学学报,2006,22,11.]doi:10.3866/PKU.WHXB20060103
(9) Li,B.;Zeng,C.G.;Li,Q.X.;Yang,J.L.;Hou,J.G.;Zhu,Q.S.J.Chin.Electr.Microsc.Soc.2003,22,189. [李 斌,曾长淦,李群祥,杨金龙,侯建国,朱清时.电子显微学报,2003,22,189.]
(10) Yourdshahyan,Y.;Zhang,H.K.;Rappe,A.M.Phys.Rev.B 2001,63,081405.doi:10.1103/PhysRevB.63.081405
(11) Vericat,C.;Vela,M.E.;Salvarezza,R.C.Phys.Chem.Chem.Phys.2005,7,3258.doi:10.1039/b505903h
(12) Maksymovych,P.;Yates,J.T.J.Am.Chem.Soc.2006,128,10642.doi:10.1021/ja062006f
(13) Maksymovych,P.;Sorescu,D.C.;Yates,J.T.J.Phys.Chem.B 2006,110,21161.doi:10.1021/jp0625964
(14) Maksymovych,P.;Vocnyy,O.;Dougherty,D.B.;Sorescu,D.C.;Yates,J.T.Pro.Surf.Sci.2010,85,206.doi:10.1016/j.progsurf.2010.05.001
(15) Vericat,C.;Vela,M.E.;Benitez,G.;Carro,P.;Salvarezza,R.C.Chem.Soc.Rev.2010,39,1805.doi:10.1039/b907301a
(16) Min,J.X.;Fan,X.L.;Cheng,Q.Z.;Chi,Q.Acta Chim.Sin.2011,69,789.[闵家祥,范晓丽,程千忠,池 琼.化学学报,2011,69,789.]
(17) Carro,P.;Torres,D.;Diaz,R.;Salvarezza,R.C.;Lllas,F.J.Phys.Chem.Lett.2012,3,2159.doi:10.1021/jz300712g
(18) Li,B.;Zeng,C.G.;Li,Q.X.;Wang,B.;Yuan,L.F.;Wang,H.Q.;Yang,J.L.;Hou,J.G.;Zhu,Q.S.J.Phys.Chem.B 2003,107,972.doi:10.1021/jp0261861
(19) Wang,J.G.;Selloni,A.J.Phys.Chem.C 2007,111,12149.doi:10.1021/jp0745891
(20) Nakayz,M.;Shikishima,M.;Shibuta,M.;Hirata,N.;Eguchi,T.;Nakajima,A.ACS Nano 2012,6,8728.doi:10.1021/nn302405r
(21) Hohenberg,P.;Kohn,W.Phys.Rev.1964,136,B864.
(22) Kohn,W.;Sham,L.J.Phys.Rev.1965,140,A1133.
(23) Kresse,G.;Hafner,J.Phys.Rev.B 1993,47,558.doi:10.1103/PhysRevB.47.558
(24) Kresse,G.;Hafner,J.Phys.Rev.B 1994,49,14251.doi:10.1103/PhysRevB.49.14251
(25) Kresse,G.;Furthmüller,J.Phys.Rev.B 1996,54,11169.doi:10.1103/PhysRevB.54.11169
(26) Kresse,G.;Furthmüller,J.Comput.Mater.Sci.1996,6,15.doi:10.1016/0927-0256(96)00008-0
(27) Kresse,G.;Joubert,D.Phys.Rev.B 1999,59,1758.doi:10.1103/PhysRevB.59.1758
(28) Blochl,P.E.Phys.Rev.B 1994,50,17953.doi:10.1103/PhysRevB.50.17953
(29) Perdew,J.P.;Burke,K.;Ernzerhof,M.Phys.Rev.Lett.1996,77,3865.doi:10.1103/PhysRevLett.77.3865
(30) Nara,J.;Higai,S.;Morikawa,Y.;Ohno,T.J.Chem.Phys.2004,120,6705.doi:10.1063/1.1651064
(31) Fan,X.L.;Chi,Q.;Liu,C.;Lau,W.J.Phys.Chem.C 2012,116,1001.
(32) Lustemberg,P.G.;Martiarena,M.L.;Martínez,A.E.;Busnengo,H.F.Langmuir 2008,24,3274.doi:10.1021/la703306t
(33) Fan,X.L.;Zhang,C.;Liu,Y.;Lau,W.M.J.Phys.Chem.C 2012,116,19909.doi:10.1021/jp306812v
(34) Maksymovych,P.;Yates,J.T.J.Am.Chem.Soc.2008,130,7518.doi:10.1021/ja800577w
(35) Rajaraman,G.;Caneschi,A.;Gatteschi,D.;Totti,F.Phys.Chem.Chem.Phys.2011,13,3886.doi:10.1039/c0cp02042g
(36) Tielens,F.;Santos,E.J.Phys.Chem.C 2010,114,9444.
(37) Gottschalck,J.;Hammer,B.J.Chem.Phys.2002,116,784.
(38) Henkelman,G.;Arnaldsson,A.;Jonsson,H.Comput.Mater.Sci.2006,36,354.doi:10.1016/j.commatsci.2005.04.010
猜你喜欢
东坡赤壁诗词(2023年2期)2023-05-30 00:04:27
课堂内外(高中版)(2020年2期)2020-05-13 14:01:40
石油石化绿色低碳(2019年6期)2019-01-14 01:16:20
铜仁学院学报(2018年6期)2018-07-05 09:47:34
中国新技术新产品(2018年2期)2018-02-02 14:55:00
衡阳师范学院学报(2016年3期)2016-07-10 07:16:27
中国塑料(2016年8期)2016-06-27 06:35:02
当代化工研究(2016年7期)2016-03-20 16:22:03
中学科技(2015年8期)2015-08-08 05:41:19
浙江理工大学学报(自然科学版)(2015年5期)2015-03-01 02:53:54
