
石墨相氮化碳的化学合成及应用
2013-09-17 06:59张金水王心晨
物理化学学报 2013年9期
张金水 王 博 王心晨
(福州大学化学化工学院,光催化研究所,福州350002)
1 引言

图1 g-C3N4可能的两种化学结构7Fig.1 Two kinds of chemical structure proposed for g-C3N47
氮化碳(C3N4)作为一种古老的聚合物,具有密度低、化学稳定性高、生物兼容性好、耐磨性强等优点,在高性能耐磨涂层、膜材料、催化剂及催化剂载体、金属氮化物的制备等领域具有广阔的应用前景,长期以来受到人们的广泛关注.1,2自从1830年Berzelius和Liebig3合成出氮化碳高分子衍生物“melon”以来,科学家们围绕氮化碳化合物的合成、表征、理论计算和应用开展了大量的研究工作.例如,1922年Franklin4通过热解Hg(CN)2和Hg(SCN)2等前驱物得到一种无定形的氮化碳,并认为它有可能进一步转变成一种更硬的相;1989年,Liu和Cohen5以β-Si3N4晶体结构为模型,采用C替代Si进行理论计算发现β-C3N4具有与金刚石相媲美的硬度和更优良的导热性能;在此基础上,1996年Teter和Hemley6采用第一性原理对C3N4进行重新计算,提出C3N4具有5种结构,即α相、β相、c相(立方相)、p相(准立方)和g相(石墨相).在这些同素异形体中,前四种都是超硬材料,具有比金刚石更高的化学惰性和热稳定性,而g-C3N4则是软质相,在常温常压下最稳定.近年来,由于超硬材料的制备要求较高,人们对氮化碳材料的研究主要集中在g-C3N4,希望通过对g-C3N4的深入研究来加深对氮化碳的了解和认识,促进g-C3N4材料科学的发展.6
如图1所示,g-C3N4可能存在两种化学结构,其中以三嗪环(C3N3)为结构单元的g1-C3N4属于R3m空间群,以七嗪环(C6N7)为结构单元的g2-C3N4属于P6m2空间群.7在这两种结构中,C、N原子均发生sp2杂化,通过pz轨道上的孤对电子形成一个类似于苯环结构的大π键,组成一个高度离域的共轭体系.在g1-C3N4中,每个三嗪环通过末端的N原子相连形成一个无限扩展的平面网格结构.其中,环内的C―N键长0.1315 nm,C―N―C键角为116.5°;环外的C―N键长0.1444 nm,C―N―C键角为116.5°.在g2-C3N4中,则用七嗪环替代三嗪环以相同的连接方式构筑C3N4.其中,环内的C―N键长0.1316 nm,C―N―C键角为116.6°;环外的C―N键长0.1442 nm,C―N―C―键角为120.0°.在这两种同素异形体中,由于结构中氮孔大小的差异导致电子结构的不同,理论上Kroke等8认为g2-C3N4更稳定,其热力学能量比 g1-C3N4低约 30 kJ·mol−1.但在实际反应中,g-C3N4的结构与原料的选择和制备方法的采用密切相关.
本论文将围绕材料的制备方法和实际应用两个方面对g-C3N4的研究现状和发展趋势作一简要概述.
2 g-C3N4的制备
目前,g-C3N4的制备方法主要分为物理法和化学法.1,2,9−11物理法主要有离子注入、反应溅射、激光束溅射等方法是早期人们在探索合成α、β、c等超硬C3N4过程中发展起来的,主要用于制备无定形的g-C3N4薄膜.9,11此外,采用机械球磨法,以三聚氰氯和氮化锂为原料可以大批量制备g-C3N4粉末.12制备g-C3N4的化学法主要包括固相反应法、溶剂热法、电化学沉积和热聚合法等,2,13具体所述如下.
2.1 固相反应法
固相反应法制备g-C3N4一般选择含有三嗪结构的化合物,如三聚氰氯、三聚氰胺等作为反应前驱体,因为三嗪结构的存在可以有效降低碳氮成键的反应能垒和促进类石墨层状晶体结构的生长.例如,Khabashesku等14以三聚氰氯为前驱物,LiN3为氮源,在300−380°C温度范围内进行固相反应,得到C/N摩尔比为0.752的无定形g-C3N4.在此基础上,Zhang等15选择三聚氰胺替代氮化锂作氮源,与三聚氰氯发生化学反应,在500−600°C,1−1.5 GPa条件下成功制备了结晶度非常好的g-C3N4,其C/N比值为0.68,但结构中含有7%(原子分数)的Cl;Gu等16以氰氨化钙作氮源,与三聚氰氯发生固相反应也能得到结晶的g-C3N4,但可惜的是样品纯度较低,含有大量的石墨碳;Guo等17通过选择NaNH2或NaN3或K与三聚氰氯在220−380°C下反应,有效调节了g-C3N4的N含量,得到C/N摩尔比分别为0.8、1.04和4的结晶g-C3N4.
Khabashesku等18还发现固相反应法可以控制材料的纳米结构,合成出具有特殊形貌的g-C3N4.他们继续以三聚氰氯和LiN3为原料,通过升高反应温度(500°C)和优化其它反应条件,得到具有空心球结构的无定形g-C3N4,其C/N比值较高,达到0.8(图2).受此启发,Lu等19采用Zn粉作催化剂,在220°C和40 MPa压力下催化三聚氰氯与NaN3发生的聚合反应,得到直径在3−6 nm,长度在100−200 nm的g-C3N4结晶纳米线.Li等20选择三聚氰氯和三聚氰胺为原料,在负压下进行高温固相反应(800°C)得到具有空心结构的g-C3N4纳米棒,其C/N比值为0.60.

图2 球状g-C3N4纳米结构的扫描电镜(SEM)(a)和透射电镜(TEM)(b)照片18Fig.2 Scanning electron microscopy(SEM)(a)and transmission electron microscopy(TEM)(b)images of spherical g-C3N4nanostructures18
以上这些固相反应法制备的g-C3N4都是基于三嗪环结构单元.最近,Komatsu21以NH4SCN为起始物,经过多步化学反应得到含有七嗪结构的化合物C6N7(NCNK)3和C6N7Cl3,并以它们为前驱物,通过高温固相法得到以七嗪环为结构单元、结晶度较好的g-C3N4,其C/N比值达到0.71,接近于g-C3N4的理论值0.75;Tragl等22则以三聚氰胺为原料,Li2(CN2)或Li3(BN2)等为氮源,50−350°C下高温固相反应得到包含三嗪环和七嗪环两种结构单元的g-C3N4,其C/N比值约0.75.
2.2 溶剂热法
溶剂热法制备C3N4具有反应条件温和、过程易于控制和体系均匀性好、流动性佳等优点.23早期,人们发展溶剂热法主要用于探索合成α相或β相超硬C3N4.例如,Zhu课题组24−26以三聚氰氯和Li3N为原料,苯为溶剂,通过改变反应温度、时间和压力,制备了一系列具有α相或β相的C3N4纳米晶.近年来,人们逐渐把溶剂热的重心转移到制备g-C3N4,探索不同前驱物制备g-C3N4的可行性.例如,Montigaud等27以三聚氰胺为原料,NH2NH2为溶剂,在800−850°C.3 GPa条件下大批量合成g-C3N4,并通过X射线衍射(XRD)、傅里叶变换红外(FT-IR)光谱和X射线光电子能谱(XPS)等对其组成结构进行详细表征.接着,他们又以三聚氰胺和三聚氰氯为原料,Et3N为溶剂,在250°C、130 MPa条件下合成结晶度较差的g-C3N4,说明样品的结晶度与反应条件,如原料、温度或压力等密切相关23.Andreyev等28以六氯苯作碳源和溶剂,NaN3为氮源,在400−500°C,7.7 GPa条下反应50−70 h得到C/N摩尔比为0.80的g-C3N4.X射线衍射(XRD)和透射电镜(TEM)等表征结果证实所制备的g-C3N4以无定形为主,包含一些结晶的纳米颗粒.Bai等29则选择简单的、常见的化合物CCl4和NH4Cl为碳源和氮源,在400°C下反应20 h,成功合成出结晶度较好的g-C3N4纳米晶,大大丰富了人们对反应前驱物的选择.
溶剂热法的另一显著优点是方便人们通过改变反应条件,控制反应物分子的自组装过程,制备出具有特殊形貌的g-C3N4的纳米结构.Xie课题组30以苯作溶剂,通过改变前驱物的组合和优化反应温度、时间和压力,成功地制备出一系列具有不同表面形貌的g-C3N4纳米材料.例如,他们以三聚氰氯和NaNH2为原料,180−220 °C下反应8−12 h得到结晶度较好的g-C3N4纳米颗粒,其中包含少量的空心球和空心立方体.他们继续优化前驱物的组合,以NaN3替代NaNH2作氮源,在220 °C下反应15 h得到内径在50−100 nm,壁厚在20−50 nm的端口封闭的g-C3N4纳米管.31此外,他们还以NH4Cl替代NaNH2作氮源,Fe作催化剂,经过220°C、15 h的反应,得到了结构非常均匀的g-C3N4纳米管,并重点研究了所制备样品的光致荧光特性.32与此同时,国内外其他课题组也纷纷开展溶剂热制备g-C3N4纳米材料的研究工作.例如,Zhu课题组33,34在一维纳米材料制备方面进行了许多尝试.他们以三聚氰氯和金属Na为前驱物,环己烷为溶剂,NiCl2为催化剂,通过优化反应温度、压力、时间等成功制备出g-C3N4纳米管、纳米带、纳米花等.此外,他们还以二聚氰胺或三聚氰胺为原料,CCl4作溶剂,在290 °C,4.5−5 MPa下反应24 h,得到结晶较好的g-C3N4纳米带和纳米管.35其中,纳米带的宽度为100 nm−3 μm,厚度为5−50 nm,长度则达到几微米到几十微米;纳米管的内壁仅5−20 nm,外壁达到70−200 nm,长度也达到几微米.最近,Wang课题组36以三聚氰氯和三聚氰胺为原料,通过筛选溶剂的种类(乙腈、苯、CCl4)和优化反应温度(140−180°C),在无催化剂存在下,制备了结构均匀的g-C3N4纳米棒(图3),并研究了其光催化分解水和光催化降解有机染料分子的反应性能.
在反应溶剂中加入SiO2等硬模板剂也可以辅助制备g-C3N4纳米结构.Bai等37在三聚氰胺与CCl4的反应体系中,添加了一定质量的80−100 nm的SiO2小球,经过250 °C、4−5 MPa、24 h溶剂热反应和20%HF除去模板剂之后得到直径在130−150 nm的g-C3N4空心球.遗憾的是,这种结构空心结构稳定性较差,去除模板剂之后,容易发生结构坍塌,大大限制了其在催化、医学等方面的应用.

图3 g-C3N4纳米棒的SEM(a)和TEM(b)照片36Fig.3 SEM(a)and TEM(b)images of g-C3N4nanorods36
2.3 电化学沉积法
电化学沉积技术不仅设备简单、控制容易,而且还能有效降低C―N成键反应能垒和降低反应体系温度,近年来开始应用于g-C3N4薄膜的制备.例如,Cao课题组38,39以1:1.5(摩尔比)的三聚氰氯和三聚氰胺乙腈饱和溶液作沉积液,首次在Si衬底上电化学沉积得到含有g-C3N4晶体结构的CNx薄膜,其C/N摩尔比值为1.25.在此基础上,他们通过改变三聚氰氯和三聚氰胺的摩尔比(如1:1和1:2),研究原料组分的变化对g-C3N4化学组成和晶体结构的影响.研究发现,当前驱物的摩尔比为1:2时,g-C3N4的结晶度较好,但样品中的N含量较低,C/N摩尔比值达到1.23.40,41最近,他们将模板法与电化学沉积法相结合,预先在ITO电极上修饰一层直径640 nm的SiO2纳米球,然后以二聚氰胺为原料,丙酮为溶剂,电化学沉积制备了直径在800 nm−1.1 μm,厚度在80−250 nm,由5−30 nm纳米颗粒组成的g-C3N4空心球(图4).42
2.4 热聚合法
热聚合法通过热诱导前驱物发生缩聚反应形成g-C3N4,是一种直接而简便的材料制备方法,近年来逐渐成为制备g-C3N4的一种常用和重要的合成方法,广泛应用于制备g-C3N4基催化剂、催化剂载体和储能材料等.2,43,44
采用热聚合法制备g-C3N4,一般得到两种类型的样品.一种是Carbon-rich g-C3N4,其C/N摩尔比值在1−5;1,45另外一种是Nitrogen-rich g-C3N4,其C/N比值在0.6−1,与g-C3N4的理论值0.75较为接近.2,43实验结果表明:Carbon-rich g-C3N4主要以乙二胺和CCl4的低温聚合物(~90°C)为起始物,经过高温(~600°C)热缩聚而成.样品中含有大量的石墨碳,结晶度较差,XRD谱图中一般只出现一个位于约26°,归属于(002)晶面的衍射峰.1,45−47Nitrogen-rich g-C3N4一般由含有三嗪结构的化合物(三聚氰胺、三聚硫氰酸)或通过低温加聚可以生成三嗪结构的化合物(氰胺、二聚氰胺、尿素)经过复杂的热聚合反应形成,聚合度较高.2,43
最近,Antonietti等2,48通过差示扫描量热法(DSC)和态密泛函理论(DFT)计算,详细研究了氰胺分子的热聚合过程.如图5所示,在60−300°C,单氰胺(CA)加热聚生成二聚氰胺(DCDA)或三聚氰胺中间体.当温度达到350°C时,中间体发生热分解,通过脱氨缩聚形成三聚氰胺二聚体(melam).继续升高温度,大约390°C时,melam进一步脱氨重排形成g-C3N4的结构基元3-s-三嗪(melem).500°C时,这种结构基元开始脱氨生成聚合度较低的高分子化合物melon.当温度高于520°C时,melon进一步缩聚得到类石墨层状结构的g-C3N4.需指出的是,g-C3N4的合成是一个复杂的热化学反应过程,不同程度的缩聚物能够在较宽的温度范围内共存,制备仅含有一种分子结构的氮化碳聚合物非常困难.当温度高于600°C时材料不稳定,发生轻微的分解,继续升高温度到700°C时,材料开始剧烈分解,生成NH3和CxNyHz等气体.因此,一般选取550−600 °C为g-C3N4的最佳合成温度.
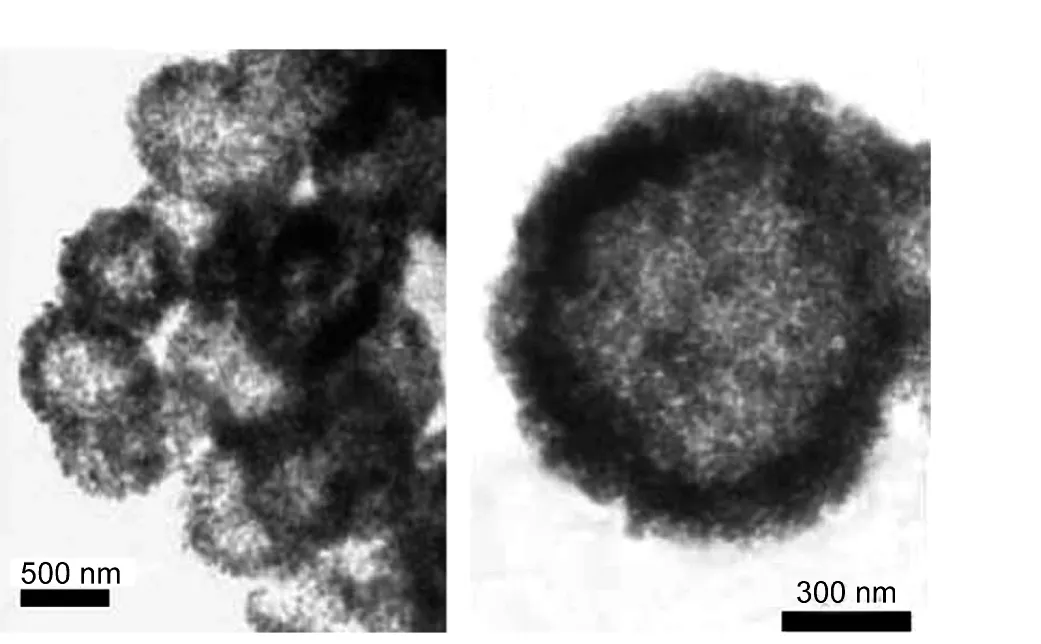
图4 g-C3N4微孔球的TEM照片42Fig.4 TEM images of g-C3N4microspheres42
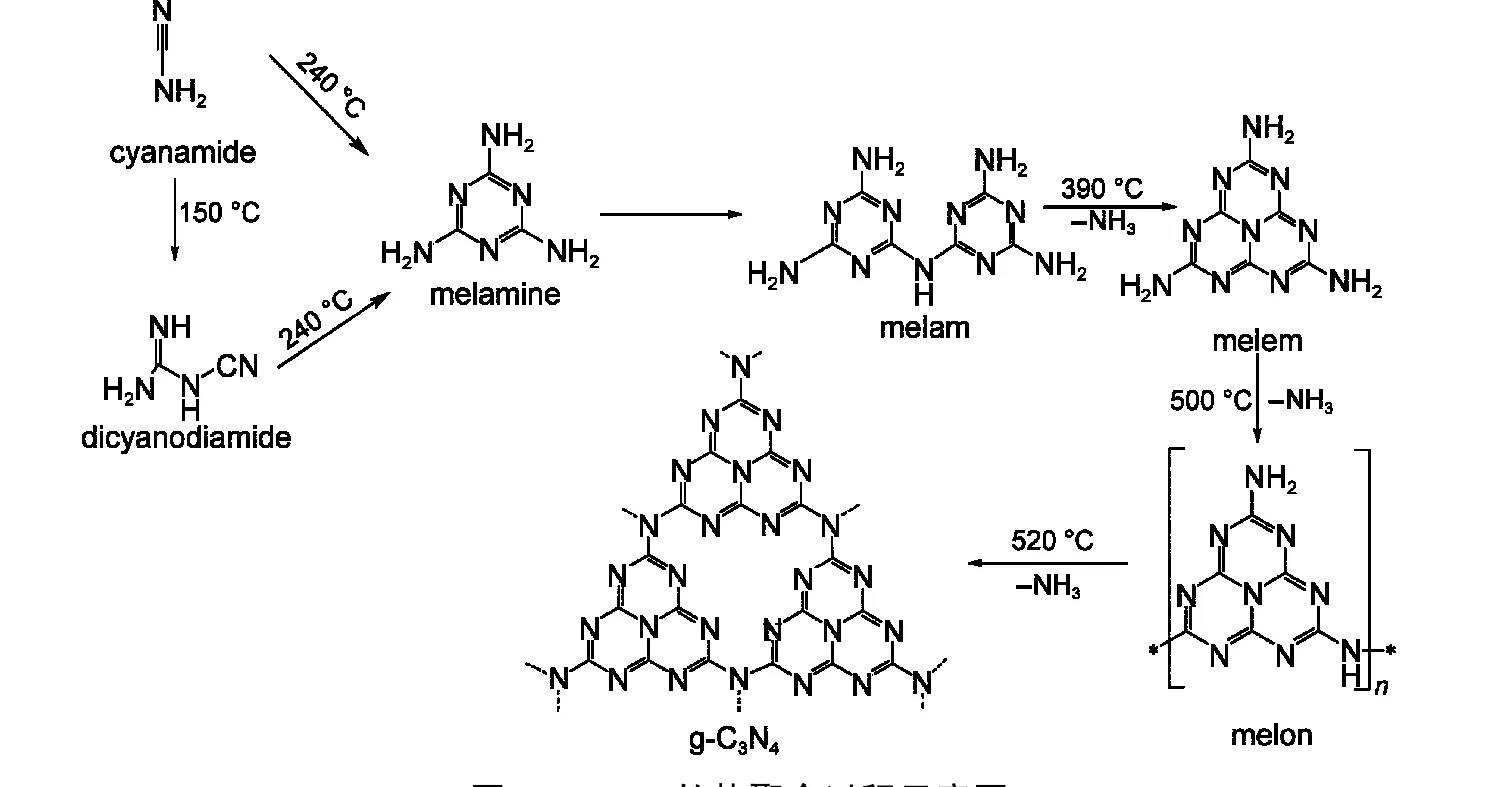
图5 g-C3N4的热聚合过程示意图Fig.5 Schematic illustration of the polymerization of g-C3N4
在热聚合法的基础上,人们还借助各种软/硬模板构筑复杂的纳米多级结构,合成出具有特殊形貌的g-C3N4.例如,采用P123作软模板剂,以二聚氰胺或三聚氰胺为前驱物,经过高温煅烧可以制备介孔g-C3N4,其比表面积最高可达300 m2·g−1;49,50在具有液态特性的前驱物,如乙二胺和CCl4的低聚物或氰胺、二聚氰胺水溶液中,添加种类丰富的SiO2,阳极氧化铝(AAO)和蒙脱土等硬模板剂,进行反向复制可以得到具有特殊形貌的g-C3N4纳米多级结构,成为制备g-C3N4纳米材料及其微观结构调控的一个研究热点.51−56
3 g-C3N4的应用
g-C3N4独特的类石墨层状堆积结构和sp2杂化的π共轭电子能带结构,使其具有多种优异的物理和化学性质,在材料、催化、电子和光学等领域具有诱人的应用前景,引起人们的广泛关注和极大兴趣.本节将就g-C3N4的应用做一个简要的介绍.
3.1 反应前驱物
Teter和Hemley6的理论计算指出,由g-C3N4向c-C3N4转变只需要12 GPa的压力,因此g-C3N4可以成为制备超硬C3N4的一种理想前驱体.当前,人们正致力于制备纯相的g-C3N4晶体,以期在真正意义上合成出氮化碳超硬材料.
此外,由于g-C3N4含有丰富的氮(~60%(w)),经常被选为含氮前驱物,用于金属氮(氧)化合物纳米结构的制备.例如,Fischer等57以介孔氮化碳(mpg-C3N4)为氮源,将金属前驱物负载在mpg-C3N4的纳米孔道内,在800°C高温煅烧制备了TiN、VN和GaN纳米粒子,证实g-C3N4既能作为氮源,又可作为模板剂,用于金属氮(氧)化物的制备;采用相同的方法,他们还制备了Al-Ga-N和Ti-V-N三元金属氮化物纳米颗粒.58在此基础上,Jun等55通过控制g-C3N4的形貌(如SBA-15型g-C3N4),成功制备了具有特殊形貌的金属氮(氧)化合物(SBA-15型TiN/C,图6).最近,Domen课题组59以mpg-C3N4为氮源,制备了具有高可见光活性的金属氮化合物Ta3N5纳米光催化剂.
3.2 催化剂载体和储能材料
g-C3N4纳米材料由于具有优异的化学惰性(pH=0−14)、较高的比表面积和种类丰富的纳米多级结构,在传统催化领域经常被用作绿色载体和储能材料.

图6 SBA-15型g-C3N4和TiN/C的TEM照片55Fig.6 TEM images of SBA-15 type g-C3N4and TiN/C55
Kim等60以g-C3N4纳米材料替代商品化的Vulcan XC-72作载体,用于负载Pt50-Ru50作甲醇燃料电池的阳极材料,结果发现电池的能量密度比原来提高了73%−83%,体现出氮化碳优异的载体效应.Datta等61利用有序介孔氮化碳(ompg-C3N4)表面丰富的胺基和孔道的纳米结构控制Au的生长,制备出Au/ompg-C3N4用于苯甲醛、六氢吡啶和苯乙炔的偶联催化反应(图7).Singh62和Zhu63等则将金负载在g-C3N4和TiO2表面,制备了Au/g-C3N4和Au/TiO2,研究催化剂表面羟基的多寡对低温氧化CO的催化性能影响.Wang等64−66将Pd负载在mpg-C3N4载体上,用于苯酚、喹啉和胺类化合物的选择性加氢,表现出很高的活性和活性选择性.Krishna等67制备g-C3N4纳米棒阵列,并将其作为载体负载Pt、Au和Fe2O3等纳米颗粒,用于硝基苯类化合物的加氢反应.
g-C3N4纳米材料表面呈弱碱性并具有较大的比表面积,在实际应用中,经常被作为一种绿色的储能材料用于H2、CO2的存储和有机物分子的吸附等.如Zhao课题组53以SBA-15、SBA-16介孔硅为模板,液态氰胺为前驱物,制备了具有二维(2D)和三维(3D)纳米结构的ompg-C3N4,并详细研究了其储氢性能,发现2D ompg-C3N4由于具有较大的比表面积,表现出更好的储氢性能,在77 K下达到1.71%(w).此外,他们还以多孔硅球MCFs为模板,乙二胺和CCl4为原料,制备了比表面积高达550 m2·g−1的氮化碳纳米球用于CO2吸附能力,使其在25和75°C下的CO2吸附量分别达到2.90和0.97 mmol·g−1.46最近,Haque等68还将2D ompg-C3N4应用于水中污染物苯酚的净化,发现ompg-CN表面碱性的―NH、―NH2基团和高度有序的介孔结构,对苯酚的高效吸附具有重要的贡献.当温度为45°C时,苯酚的吸附量达到769 mg·g−1.
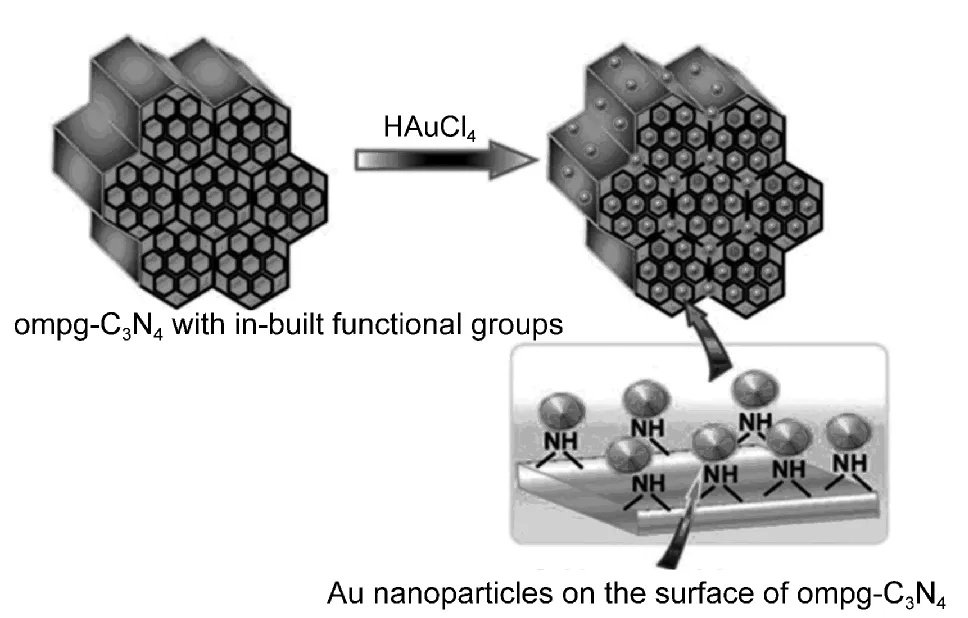
图7 在无其它稳定剂时利用ompg-C3N4的内建官能团自组装Au纳米颗粒61Fig.7 Encapsulation ofAu nanoparticles over ompg-C3N4 with in-built functional groups without any external stabilizing agent61
3.3 传感器
g-C3N4有机半导体由于表面含有大量的氨基,近年来开始作为传感器用于金属离子的监测、酸性气体的检测和生物成像等方面.例如,Jun课题组69将3D ompg-C3N4作为荧光传感器,用于检测溶液中痕量的金属离子,发现其对Cu2+具有很好的选择性和很高的灵敏度.在此基础上,他们以Cu2+-ompg-C3N4组合作为传感器,用于检测水溶液和人体血液中CN−的浓度,发现CN−的检出限低至80 nmol·L−1.70Cheng等71详细研究了g-C3N4电化学发光特性,并将其用于电致发光检测溶液中的Cu2+,拓展了g-C3N4在电化学检测方面的应用(图8).Mane等72将2D ompg-C3N4与石英微天平组合作为气体传感器,用于检测空气中酸性和碱性的有机气体,由于2D ompg-C3N4表面有大量的氨基呈弱碱性,发现传感器对甲酸气体的响应最为灵敏,选择性最高.最近,Xie课题组73将g-C3N4进行溶液剥层,制备出具有水溶性的g-C3N4纳米片,发现其荧光强度较体相材料提高了~20%,可以作为荧光材料,用于生物成像.
3.4 氧还原催化剂
阴极上氧还原反应对于燃料电池的性能具有非常重要的作用.目前,最高效的氧还原催化剂是Pt,但Pt昂贵的价格和易失活的缺点,极大地限制了燃料电池的商业化应用.因此,开发Pt替代材料成为燃料电池研究的一个中心任务.N掺杂C由于出色的氧还原能力,已经被作为一种非金属氧还原材料广泛应用于燃料电池的研究中.74最近,g-C3N4由于类石墨层状结构特点,也被作为一种氧还原材料广泛应用于燃料电池的研究.44,74
如图9所示,Lyth等75首次发现g-C3N4具有氧还原能力,在酸性介质中其催化活性明显高于炭黑.但g-C3N4材料较差的导电能力(108−1010Ω)和较低的比表面积(~10 m2·g−1),严重抑制了其氧还原的能力,使反应电流密度和还原起始电位远小于商品化的Pt/C,制约了g-C3N4在燃料电池中的应用.在此基础上,他们发现通过提高煅烧温度(~1000°C)或使用炭黑(CB)作导电载体,可以在一定程度上降低g-C3N4氧还原的过电位和提高电流密度,为g-C3N4材料的改性研究指明了方向.76Kwon等77在体相g-C3N4中引入有序介孔结构,有效地扩大材料的比表面积,从而显著提高其氧还原能力;Yang等78通过表面自组装的方法,在石墨烯氧化物(GO)的表面生长g-C3N4,形成一种类三明治的G-g-C3N4纳米层状薄片,有效地克服了g-C3N4比表面积小、导电能力差等缺点,使G-g-C3N4的氧还原能力与商品化的Pt/C相媲美,并表现出更好的活性稳定性.Sun等79采用溶液法制备的氮化碳石墨烯复合材料也表现出相似的氧还原特性.最近,Qiao课题组80,81分别采用2D的有序介孔炭和3D的微孔炭球作载体负载g-C3N4,显著提高材料的比表面积和导电能力,从而有效地克服氧还原反应中的动力学问题,提高反应过程中的4电子转移过程.
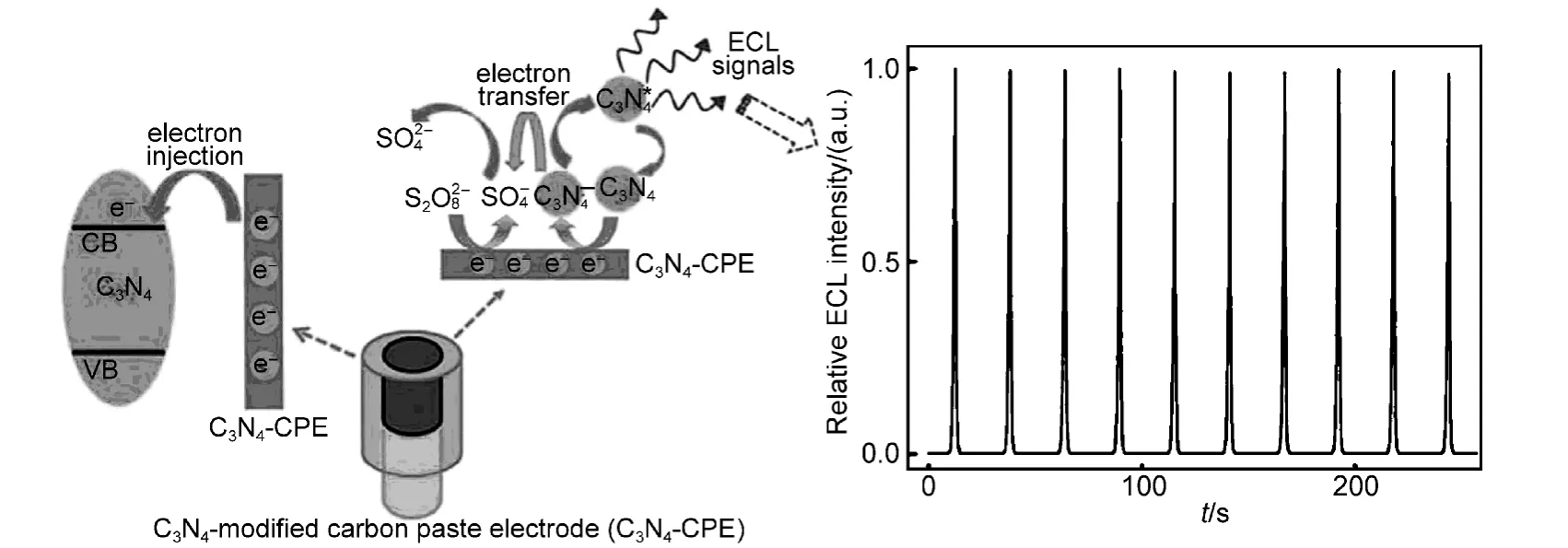
图8 g-C3N4电化学发光检测Cu2+示意图71Fig.8 Schematic illustration of the electroluminescence determination of Cu2+on g-C3N471

图9 g-C3N4的氧还原性质75Fig.9 Oxygen reduction reaction property of g-C3N475
3.5 有机反应催化剂
如图10所示,g-C3N4由于独特的化学组成和π共轭电子结构,具有较强的亲核能力、易形成氢键以及Brönsted碱功能和Lewis碱功能,使其可以成为一种多功能的催化剂,应用于传统的有机催化反应中.2,43
Antonietti课题组54将mpg-C3N4作为不含金属组分的Lewis碱催化剂,用于活化苯环上的C―N键,使其与己酰氯发生Friedel-Crafts酰化反应.他们不仅考察了催化剂的比表面积、孔结构和聚合度对Friedel-Crafts酰化反应的影响,而且还使用甲醇、乙醇和甲酸等不含氯的电子亲核试剂替代己酰氯与苯发生Friedel-Crafts反应,开拓了g-C3N4在绿色化学合成中的重要应用.82Vinu51则以乙二胺和CCl4为原料,SBA-15介孔硅为模板合成了2D ompg-C3N4,并将其用于苯和己酰氯的Friedel-Crafts酰化反应.此外,mpg-C3N4还可以活化各种双键和三键,使其发生Pauson-Khand反应,催化各种腈类和炔化合物发生成环反应.83
利用mpg-C3N4Lewis碱上的孤对电子和表面残留的―NH,很容易通过形成氨基甲酸酯六元环进行有效地固定CO2,而且还可以通过Lewis碱活化苯,使其和催化剂表面固定的CO2发生氧化还原反应,生成苯酚和CO,实现CO2的绿色固定.84此外,Ansari等85继续以mpg-C3N4为催化剂,活化CO2使其与环氧丙烷发生环加成反应,生成相应的五元环化合物.
最近,Vinu47和Wang87课题组采用g-C3N4替代一些常用的无机或有机碱,用于传统的有机碱催化反应中.例如,将SBA-15型ompg-C3N4用于催化各种醇类,形成β-酮酸酯(β-keto Ester);用mpg-C3N4或具有3D结构的ompg-C3N4催化苯甲醛与丙二腈发生Knoevenagel缩合反应,得到α,β-不饱和羰基化合物,具有很高的选择性.而且使用K2CO3,KOH等对其进行碱化处理,还可进一步提高其催化转化效率.86,87
g-C3N4由于离域的π共轭电子结构,可以高效活化H2O2或O2,从而拓展其在有机选择性氧化中的应用.Antonietti课题组88以BmimBF4离子液和二聚氰胺为原料,制备了B-F原位共掺杂的介孔氮化碳,并以此为催化剂,活化H2O2,实现了环己烷到环己酮的转化,转化率和选择性分别为10.1%和43%,明显高于传统的g-C3N4.在分子氧活化方面,他们以mpg-C3N4和硼掺杂氮化碳为催化剂,O2为氧化剂,实现了饱和C―H键的活化,分别成功地将甲苯和二苯基甲烷转化为苯甲醛或二苯甲酮.89,90此外,他们还采用g-C3N4和石墨烯复合物活化分子氧,实现了饱和C―H键的高效转化,并探讨了相关的反应机理.91
3.6 光催化剂
g-C3N4是一种典型的聚合物半导体,结构中的C、N原子以sp2杂化形成高度离域的π共轭体系.Wang及其合作者92,93借助态泛函理论详细研究了其半导体能带结构,发现其最高已占据轨道(HOMO)由N pz轨道组成,最低未占据轨道(LUMO)由C pz组成,它们之间存在2.6 eV的禁带宽度,可以吸收太阳光谱中波长小于475 nm蓝紫光(图11).更为重要的是,g-C3N4的HOMO和LUMO带边位置分别在+1.6和−1.0 V(vs)NHE(标准氢电极),较为合适,在热力学上可以分解水产氢、产氧.其中HOMO位置在1.6 V(vs NHE),比φO2/H2O=1.23 V电极电位低,说明光生空穴可以氧化水生成氧气;LUMO位置在−1.0 V(vs NHE),高于φH+/H2=0 V,表明光生电子具有很强的还原能力,可以还原水产生氢气.所以,理论上g-C3N4可以作为一种具有可见光响应的光催化材料,应用于光催化领域.
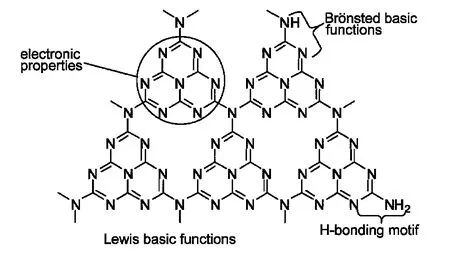
图10 g-C3N4与化学结构有关的催化反应特性2,43Fig.10 Structure-dependence chemical properties of g-C3N4as a catalyst2,43
在此基础上,Wang及其合作者92−101以三乙醇胺为电子给体、Pt纳米颗粒为产氢助催化剂或以Ag-NO3为电子牺牲剂、RuO2为产氧助催化剂、La2O3为溶液缓冲剂,分别考察了传统g-C3N4及其改性修饰后样品的光解水产氢、产氧半反应性能.发现在可见光(λ>420 nm)照射下,g-C3N4可持续产生氢气或氧气,将g-C3N4作为一种不含金属组分的聚合物半导体引入到光催化领域(图12).到目前为止,g-C3N4作为一种可见光催化剂,被广泛应用于能源光催化、环境光催化、有机选择性光合成和光催化聚合物的制备等方面,受到人们的广泛关注.102,103
例如,Wang及其合作者104以卟啉铁、酞箐铁等结构为基础,仿生合成了Fe掺杂的g-C3N4(Fe-g-C3N4),发现其具有很好的光催化活化H2O2、降解RhB的性能,将g-C3N4应用于水相中有机污染物的光催化降解.此后,Zou课题组105以三聚氰胺为原料,制备g-C3N4,并将其作为不含金属组分的光催化材料用于MO的可见光降解,进一步推动g-C3N4在环境光催化领域中的应用.
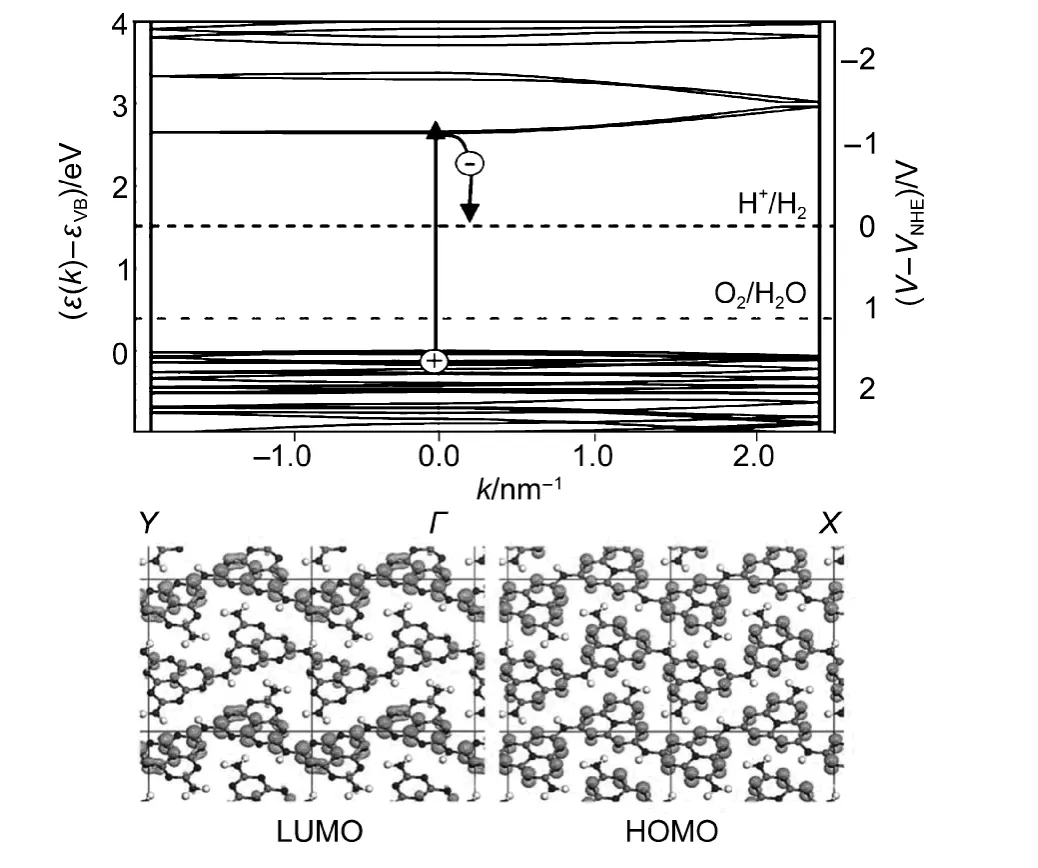
图11 聚合物melon的电子结构92Fig.11 Electronic structures of polymeric melon92
有机选择性光合成方面,Wang及其合作者106将Fe-g-C3N4负载在SBA-15载体上,用于光催化活化H2O2,实现苯到苯酚的高效转化,在4 h中苯的转化率达到11.9%,生成苯酚的TOF(每摩尔催化剂上苯酚的转化频率)达到1484.1 h−1.接着,他们又继续报道在温和条件下,mpg-C3N4可以光催化活化分子氧生成超氧自由基,同时又避免产生具有强氧化能力的羟基自由基,帮助人们控制有机分子的光催化氧化过程,实现反应底物的光催化定向转化.例如,可以将醇类化合物定向光催化脱氢氧化生成相应的醛/酮;107或将胺类化合物高效和高选择性光催化氧化偶联合成亚胺.108此外,Möhlmann109和Zhang110,111等还将mpg-C3N4应用于C―C键的光催化活化和有机硫化物的选择性合成.
图12 g-C3N4的光解水产氢、产氧活性92Fig.12 Photocatalytic production of H2and O2on g
最近,Wang和Yagci等112合作,进一步开发g-C3N4材料在光催化领域中的应用,将其拓展到功能性聚合物的光催化合成中.以mpg-C3N4为光催化剂,在可见光照下,利用光生空穴活化三乙胺,产生三乙胺自由基,驱动丙烯酸甲酯发生聚合反应,生成聚丙烯酸甲酯.这一研究有效地推动了g-C3N4在聚合物制备领域的新应用.
现在,g-C3N4作为一种廉价、稳定、具有良好可见光响应的聚合物半导体光催化剂,越来越受到人们的广泛关注.
4 结论与展望
到目前为止,人们已经开发了固相反应法、溶剂热法、电化学沉积法和热聚合法等多种制备g-C3N4聚合物半导体的化学方法,大大促进了g-C3N4材料科学的发展.其中,热聚合法由于操作简单、安全、可以大批量合成和反应条件易控制等优点,正逐渐成为g-C3N4制备的主要方法.103在此基础上,人们积极拓展g-C3N4在能源和材料相关领域的重要应用,发现g-C3N4可以作为不含金属组分的催化剂和催化剂载体广泛应用于有机官能团的选择性转换、光催化分解水、氧化原和Au、Pd、Ag、Pt等贵金属的负载,或作为绿色储能材料和硬模板剂用于H2、CO2的存储和金属氮(氧)化物的制备等,引起了人们的重视.
今后,g-C3N4学科的发展仍将围绕材料的制备和应用研究展开.在材料合成方面,人们将会把更多的精力集中到g-C3N4材料的可控制备上来,采用各种途径和手段优化g-C3N4的化学组成、调控其半导体能带结构和表面形貌等,按照特定的实验目的对g-C3N4材料进行改性研究.在g-C3N4的应用方面,由于能源短缺和环境危机日益严重,人们将会把主要的精力投入到g-C3N4光催化、氧化原和有机选择性合成等研究中,并进一步拓展g-C3N4在能源和环境领域的应用.
(1) Kroke,E.;Schwar,M.Coord.Chem.Rev.2004,248,493.doi:10.1016/j.ccr.2004.02.001
(2) Thomas,A.;Fischer,A.;Goettmann,F.;Antonietti,M.;Müller,J.;Schlögl,R.;Carlsson,J.J.Mater.Chem.2008,18,4893.doi:10.1039/b800274f
(3) Liebig,J.Ann.Pharm.1834,10,10.
(4) Franklin,E.J.Am.Chem.Soc.1922,44,486.doi:10.1021/ja01424a007
(5) Liu,A.;Cohen,M.Science 1989,245,841.doi:10.1126/science.245.4920.841
(6) Teter,D.;Hemley,R.Science 1996,271,53.doi:10.1126/science.271.5245.53
(7) Molina,B.;Sansores,L.Modern Phys.Lett.B 1999,13,193.doi:10.1142/S0217984999000269
(8) Kroke,E.;Sehwarz,M.;Horath-Bordon,E.;Kroll,P.;Noll,B.;Norman,A.New J.Chem.2002,26,508.doi:10.1039/b111062b
(9) Chen,T.B.;Li,Z.Q.Matrer.Sci.Eng.1998,16,6.[陈天兵,李宗全.材料科学与工程,1998,16,6.]
(10) Ma,Z.New Carbon Mater.2006,21,276.[马志斌.新型炭材料,2006,21,276.]
(11)Wu,X.C.;Liu,G.H.;Chen,G.H.J.Gansu Sci.1999,11,9.[吴现成,刘国汉,陈光华.甘肃科学学报,1999,11,9.]
(12) Zhao,H.;Chen,X.;Jia,C.;Zhou,T.;Qu,X.;Jian,J.;Xu,Y.;Zhou,T.Mater.Sci.Eng.B 2005,122,90.doi:10.1016/j.mseb.2005.05.007
(13) Meng,Y.L.Study on the Preparation and Photocatalysis of Graphitic Carbon Nitrides.Master Dissertation,Dalian University of Technology,Dalian,2010.[梦雅丽.g-C3N4的合成及其光催化研究[D].大连:大连理工大学,2011.]
(14) Khabashesku,V.;Zimmerman,J.;Margrave,J.Chem.Mater.2000,12,3264.doi:10.1021/cm000328r
(15) Zhang,Z.;Leinenweber,K.;Bauer,M.;Garvie,L.;McMillan,P.;Wolf,G.J.Am.Chem.Soc.2001,123,7788.doi:10.1021/ja0103849
(16) Gu,Y.;Chen,L.;Shi,L.;Ma,J.;Yang,Z.;Qian,Y.Carbon 2003,41,2653.doi:10.1016/S0008-6223(03)00287-2
(17) Guo,Q.;Yang,Q.;Yi,C.;Zhu,L.;Xie,Y.Carbon 2005,43,1386.doi:10.1016/j.carbon.2005.01.005
(18) Zimmerman,J.;Williams,R.;Khabashesku,V.;Margrave,J.Nano Lett.2001,1,731.doi:10.1021/nl015626h
(19) Lu,X.;Gai,L.;Cui,D.;Wang,Q.;Zhao,X.;Tao,X.Mater.Lett.2007,61,4255.doi:10.1016/j.matlet.2007.01.076
(20) Li,Y.;Zhang,J.;Wang,Q.;Jin,Y.;Huang,D.;Cui,Q.;Zou,G.J.Phys.Chem.B 2010,114,9429.doi:10.1021/jp103729c
(21) Komatsu,T.J.Mater.Chem.2001,11,802.doi:10.1039/b007165j
(22) Tragl,S.;Gibson,K.;Glaser,J.;Duppel,V.;Simon,A.;Meyer,H.Solid State Commun.2007,14,529.
(23) Montigaud,H.;Tanguy,B.;Demazeau,G.;Alves,I.;Courjault,S.J.Mater.Sci.2000,35,2547.doi:10.1023/A:1004798509417
(24) Lv,Q.;Cao,C.;Li,C.;Zhang,J.;Zhu,H.;Kong,X.;Duan,X.J.Mater.Chem.2003,13,1241.
(25) Luv,Q.;Cao,C.;Zhang,J.;Li,C.;Zhu,H.J.Appl.Phys.A 2004,79,633.doi:10.1007/s00339-002-2058-4
(26) Fu,Q.;Cao,C.;Zhu,H.Chem.Phys.Lett.1999,314,223.doi:10.1016/S0009-2614(99)01169-0
(27) Montigaud,H.;Tanguy,B.;Demazeau,G.;Alves,I.;Birot,M.;Dungues,J.Diam.Relat.Muter.1999,8,1707.
(28)Andreyev,A.;Akaishi,M.;Golberg,D.Chem.Phys.Lett.2003,372,635.doi:10.1016/S0009-2614(03)00471-8
(29) Bai,Y.;Lv,B.;Liu,Z.;Li,L.;Cui,D.;Xu,X;Wang,Q.J.Cryst.Growth 2003,247,505.doi:10.1016/S0022-0248(02)01981-4
(30) Guo,Q.;Xie,Y.;Wang,X.;Lv,S.;Hou,T.;Liu,X.Chem.Phys.Lett.2003,380,84.doi:10.1016/j.cplett.2003.09.009
(31) Guo,Q.;Xie,Y.;Wang,X.;Zhang,S.;Hou,T.;Lv,S.Chem.Commun.2004,26.
(32) Guo,Q.;Yang,Q.;Zhu,L.;Yi,C.;Zhang,S.;Xie,Y.Solid State Commun.2004,132,369.doi:10.1016/j.ssc.2004.08.014
(33) Cao,C.;Huang,F.;Cao,C.;Li,J.;Zhu,H.Chem.Mater.2004,16,5213.doi:10.1021/cm0493039
(34) Li,J.;Cao,C.;Hao,J.;Qiu,H.;Xu,Y.;Zhu,H.Diam.Relat.Mater.2006,15,1593.doi:10.1016/j.diamond.2006.01.013
(35) Li,J.;Cao,C.;Zhu,H.Nanotechnology 2007,18,115605.doi:10.1088/0957-4484/18/11/115605
(36) Cui,Y.;Ding,Z.;Fu,X.;Wang,X.Angew.Chem.Int.Edit.2012,51,11814.doi:10.1002/anie.201206534
(37) Bai,X.;Li,J.;Cao,C.;Hussain,S.Mater.Lett.2011,65,1101.doi:10.1016/j.matlet.2011.01.008
(38) Li,C.;Cao,C.;Zhu,H.Chin.Sci.Bull.2003,48,1737.doi:10.1360/03wb0011
(39) Li,C.;Cao,C.B.;Zhu,H.S.;Lv,Q.;Zhang,J.T.;Xiang,X.J.Synt.Cryst.2003,32,252.[李 超,曹传宝,朱鹤孙,吕 强,张加涛,项 顼.人工晶体学报,2003,32,252.]
(40) Li,C.;Cao,C.;Zhu,H.;Lv,Q;Zhang,J.Wang,X.Mater.Sci.Eng.B 2004,106,308.doi:10.1016/j.mseb.2003.10.006
(41) Li,C.;Cao,C.;Zhu,H.Mater.Lett.2004,58,1903.doi:10.1016/j.matlet.2003.11.024
(42) Bai,X.;Li,J.;Cao,C.Appl.Surf.Sci.2010,256,2327.doi:10.1016/j.apsusc.2009.10.061
(43) Wang,Y.;Wang,X.;Antonietti,M.Angew.Chem.Int.Edit.2012,51,68.doi:10.1002/anie.201101182
(44) Zheng,Y.;Liu,J.;Liang,J.;Jaroniec,M.;Qiao,S.Energy Environ.Sci.2012,5,6717.doi:10.1039/c2ee03479d
(45) Lyth,S.;Nabae,Y.;Moriya,S.;Kuroki,S.;Kakimoto,M.;Ozaki,J.;Miyata,S.J.Phys.Chem.C 2009,113,20148.doi:10.1021/jp907928j
(46) Li,Q.;Yang,J.;Feng,D.;Wu,Z.;Wu,Q.;Park,S.;Ha,C.;Zhao,D.Nano Res.2010,3,632.doi:10.1007/s12274-010-0023-7
(47) Jin,X.;Balasubramanian,V.;Selvan,S.;Sawant,D.;Chari,M.;Lu,G.;Vinu,A.Angew.Chem.Int.Edit.2009,48,7884.doi:10.1002/anie.v48:42
(48) Groenewolt,M.;Antonietti,M.Adv.Mater.2005,17,1789.
(49)Wang,Y.;Wang,X.;Antonietti,M.;Zhang,Y.ChemSusChem 2010,3,435.doi:10.1002/cssc.v3:4
(50)Yang,H.Chem.Commun.2012,48,3430.doi:10.1039/c2cc00001f
(51) Vinu,A.Adv.Funct.Mater.2008,18,816.
(52) Talapaneni,S.;Anandan,S.;Mane,G.;Anand,C.;Dhawale,D.;Varghese,S.;Mano,A.;Mori,T.;Vinu,A.J.Mater.Chem.2012,22,9831.doi:10.1039/c2jm30229b
(53) Park,S.;Chu,S.;Xue,C.;Zhao,D.;Ha,C.J.Mater.Chem.2011,21,10801.doi:10.1039/c1jm10849b
(54) Goettmann,F.;Fischer,A.;Antonietti,M.;Thomas,A.Angew.Chem.Int.Edit.2006,45,4467.
(55)Jun,Y.;Hong,W.;Antonietti,M.;Thomas,A.Adv.Mater.2009,21,4270.doi:10.1002/adma.v21:42
(56) Zhang,J.;Guo,F.;Wang,X.Adv.Funct.Mater.2013,23,3008.doi:10.1002/adfm.201203287.
(57) Fischer,A.;Antonietti,M.;Thomas,A.Adv.Mater.2007,19,264.
(58) Fischer,A.;Muller,J.;Antonietti,M.;Thomas,A.ACS Nano,2008,2,2489.doi:10.1021/nn800503a
(59) Yuliati,L.;Yang,J.;Wang,X.;Maeda,K.;Takata,T.;Antonietti,M.;Domen,K.J.Mater.Chem.2010,20,4295.doi:10.1039/c0jm00341g
(60) Kim,M.;Hwang,S.;Yu,J.J.Mater.Chem.2007,17,1656.doi:10.1039/b702213a
(61) Datta,K.;Reddy,B.;Ariga,K.;Vinu,A.Angew.Chem.Int.Edit.2010,49,5961.
(62) Singh,J.;Overbury,S.;Dudney,N.;Li,M.;Veith,G.ACS Catal.2012,2,1138.doi:10.1021/cs3001094
(63) Zhu,J.;Carabineiro,S.;Shan,D.;Faria,J.;Zhu,Y.;Figueiredo,J.J.Catal.2010,274,207.doi:10.1016/j.jcat.2010.06.018
(64) Wang,Y.;Yao,J.;Li,H.;Su,D.;Antonietti,M.J.Am.Chem.Soc.2011,133,2362.doi:10.1021/ja109856y
(65) Li,Y.;Gong,Y.;Xu,X.;Zhang,P.;Li,H.;Wang,Y.Catal.Commun.2012,28,9.doi:10.1016/j.catcom.2012.08.005
(66) Gong,Y.;Zhang,P.;Xu,X.;Li,Y.;Li,H.;Wang,Y.J.Catal.2013,297,272.doi:10.1016/j.jcat.2012.10.018
(67) Krishna,K.;Kumar,B.;Eswaramoorthy,M.Chem.Phys.Lett.2011,511,87.doi:10.1016/j.cplett.2011.06.006
(68) Haque,E.;Jun,J.;Talapaneni,S.;Vinu,A.;Jhunq,S.J.Mater.Chem.2010,20,10801.doi:10.1039/c0jm02974b
(69) Lee,E.;Jun,Y.;Hong,W.;Thomas,A.;Jin,M.Angew.Chem.Int.Edit.2010,49,9706.doi:10.1002/anie.201004975
(70) Lee,E.;Lee,S.;Heo,N.;Stucky,G.;Jun,Y.;Hong,W.Chem.Commun.2012,48,3942.doi:10.1039/c2cc17909a
(71) Cheng,C.;Huang,Y.;Tian,X.;Zheng,B.;Li,Y.;Yuan,H.;Xiao,D.;Xie,S.;Choi,M.Anal.Chem.2012,84,4754.doi:10.1021/ac300205w
(72)Mane,G.;Dhawale,D.;Anand,C.;Ariga,K.;Ji,Q.;Wahab,M.;Mori,T.;Vinu,A.J.Mater.Chem.A 2013,1,2913.doi:10.1039/c2ta01215d
(73) Zhang,X.;Xie,X.;Wang,H.;Zhang,J.;Pan,B.;Xie,Y.J.Am.Chem.Soc.2013,135,18.doi:10.1021/ja308249k
(74) Zheng,Y.;Jiao,Y.;Jaroniec,M.;Jin,Y.;Qiao,S.Small 2012,8,3550.doi:10.1002/smll.v8.23
(75) Lyth,S.;Nabae,Y.;Moriya,S.;Kuroki,S.;Kakimoto,M.;Ozaki,J.;Miyata,S.J.Phys.Chem.C 2009,113,20148.doi:10.1021/jp907928j
(76) Lyth,S.;Nabae,Y.;Islam,N.;Kuroki,S.;Kakimoto,M.;Miyata,S.J.Electrochem.Soc.2011,158,B194.
(77) Kwon,K.;Sa,Y.;Cheon,J.;Joo,S.Langmuir 2012,28,991.doi:10.1021/la204130e
(78) Yang,S.;Feng,X.;Wang,X.;Müllen,K.Angew.Chem.Int.Edit.2011,50,5339.doi:10.1002/anie.201100170
(79) Sun,Y.;Li,C.;Xu,Y.;Bai,H.;Yao,Z.;Shi,G.Chem.Commun.2010,46,4740.doi:10.1039/c001635g
(80) Zheng,Y.;Jiao,Y.;Chen,J.;Liu,J.;Liang,J.;Du,A.;Zhang,W.;Zhu,Z.;Smith,S.;Jaroniec,M.;Qiao,S.J.Am.Chem.Soc.2011,133,20116.doi:10.1021/ja209206c
(81) Liang,J.;Zheng,Y.;Chen,J.;Liu,J.;Hulicova-Jurcakova,D.Jaroniec,M.;Qiao,S.Angew.Chem.Int.Edit.2012,51,3892.doi:10.1002/anie.201107981
(82) Goettmann,F.;Fischer,A.;Antonietti,M.;Thomas,A.Chem.Commun.2006,4530.
(83) Goettmann,F.;Fischer,A.;Antonietti,M.;Thomas,A.New J.Chem.2007,31,1455.doi:10.1039/b618555j
(84) Goettmann,F.;Thoma,A.;Antonietti,M.Angew.Chem.Int.Edit.2007,46,2717.
(85)Ansari,M.;Min,B.;Mo,Y.;Park,S.Green Chem.2011,13,1416.doi:10.1039/c0gc00951b
(86) Talapaneni,S.;Anandan,S.;Mane,G.;Anand,C.;Dhawale,D.;Varghese,S.;Mano,A.;Mori,T.;Vinu,A.J.Mater.Chem.2012,22,9831.doi:10.1039/c2jm30229b
(87) Su,F.;Antonietti,M.;Wang,X.Catal.Sci.Technol.2012,2,1005.doi:10.1039/c2cy00012a
(88) Wang,Y.;Zhang,J.;Wang,X.;Antonietti,M.;Li,H.Angew.Chem.Int.Edit.2010,49,3356.doi:10.1002/anie.201000120
(89) Li,X.;Wang,X.;Antonietti,M.ACS Catal.2012,2,2082.doi:10.1021/cs300413x
(90) Wang,Y.;Li,H.;Yao,J.;Wang,X.;Antonietti,M.Chem.Sci.2011,2,446.doi:10.1039/c0sc00475h
(91) Li,X.;Chen,J.;Wang,X.;Sun,J.;Antonietti,M.J.Am.Chem.Soc.2011,133,8074.
(92) Wang,X.;Maeda,K.;Thomas,A.Nat.Mater.2009,8,76.doi:10.1038/nmat2317
(93) Maeda,K.;Wang,X.;Nishihara,Y.J.Phys.Chem.C 2009,113,4940.
(94) Zhang,J.;Chen,X.;Takanabe,K.;Maeda,K.;Domen,K.;Epping,J.;Fu,X.;Antonietti,M.;Wang,X.Angew.Chem.Int.Edit.2010,49,441.doi:10.1002/anie.200903886
(95) Zhang,J.;Zhang,G.;Chen,X.;Lin,S.;Möhlmann,L.;Dolega,G.;Lipner,G.;Antonietti,M.;Blechert,S.;Wang,X.Angew.Chem.Int.Edit.2012,51,3183.doi:10.1002/anie.v51.13
(96) Zhang,J.;Zhang,M.;Sun,R.;Wang,X.Angew.Chem.Int.Edit.2012,51,10145.doi:10.1002/anie.201205333
(97) Zhang,J.;Zhang,M.;Lin,S.;Fu,X.;Wang,X.J.Catal.2013,doi:10.1016/j.jcat.2013.01.008.
(98) Zhang,J.;Sun,J.;Maeda,K.;Domen,K.;Liu,P.;Antonietti,M.;Fu,X.;Wang,X.Energy Environ.Sci.2011,4,675.doi:10.1039/c0ee00418a
(99) Zhang,J.;Grzelczak,M.;Hou,Y.;Maeda,K.;Domen,K.;Fu,X.;Antonietti,M.;Wang,X.Chem.Sci.2012,3,443.doi:10.1039/c1sc00644d
(100) Zhang,J.;Zhang,M.;Zhang,G.;Wang,X.ACS Catal.2012,2,940.doi:10.1021/cs300167b
(101) Zheng,H.R.;Zhang,J.S.;Wang,X.C.;Fu,X.Z.Acta Phys.-Chim.Sin.2012,28,2336.[郑华荣,张金水,王心晨,付贤智.物理化学学报,2012,28,2336.]doi:10.3866/PKU.WHXB201209104
(102) Chen,X.;Shen,S.;Guo,L.;Mao,S.Chem.Rev.2010,110,6503.doi:10.1021/cr1001645
(103) Wang,X.;Blechert,S.;Antonietti,M.ACS Catal.2012,2,1596.doi:10.1021/cs300240x
(104) Wang,X.;Chen,X.;Thomas,A.;Fu,X.;Antonietti,M.Adv.Mater.2009,21,1609.doi:10.1002/adma.v21:16
(105) Yan,S.;Li,Z.;Zou,Z.Langmuir 2009,25,10397.doi:10.1021/la900923z
(106) Chen,X.;Zhang,J.;Fu,X.;Antonietti,M.;Wang,X.J.Am.Chem.Soc.2009,131,11658.doi:10.1021/ja903923s
(107) Su,F.;Mathew,S.;Lipner,G.;Fu,X.;Antonietti,M.;Blechert,S.;Wang,X.J.Am.Chem.Soc.2010,132,16299.doi:10.1021/ja102866p
(108) Su,F.;Mathew,S.;Möhlmann,L.;Antonietti,M.;Wang,X.;Blechert,S.Angew.Chem.Int.Edit.2011,50,657.doi:10.1002/anie.v50.3
(109) Möhlmann,L.;Baar,M.;Rieß,J.;Antonietti,M.;Wang,X.;Blechert,S.Adv.Synth.Catal.2012,354,1909.doi:10.1002/adsc.v354.10
(110) Zhang,P.;Wang,Y.;Li,H.;Antonietti,M.Green Chem.2012,14,1904.doi:10.1039/c2gc35148j
(111) Zhang,P.;Wang,Y.;Yao,J.Adv.Synth.Catal.2011,353,1447.doi:10.1002/adsc.201100175
(112) Kiskan,B.;Zhang,J.;Wang,X.;Antonietti,M.;Yagci,Y.ACS Macro.Lett.2012,1,546.doi:10.1021/mz300116w
猜你喜欢
陶瓷学报(2021年1期)2021-04-13
粉末冶金技术(2021年1期)2021-03-29
陶瓷学报(2020年2期)2020-10-27
热处理技术与装备(2019年1期)2019-03-14
电子制作(2018年12期)2018-08-01
中国资源综合利用(2017年4期)2018-01-22
环境与生活(2016年11期)2016-12-02
当代化工研究(2016年7期)2016-03-20
应用化工(2014年11期)2014-08-16
