
Y-39983在实验性自身免疫性脑脊髓炎中的作用研究
2013-09-17 03:58:28谢富华张姗姗林哲聪高聪熊旭明
中国神经免疫学和神经病学杂志 2013年2期
谢富华 张姗姗 林哲聪 高聪 熊旭明
多发性硬化(multiple sclerosis,MS)是具有轴突损害的一类脱髓鞘性疾病,实验性自身免疫性脑脊 髓 炎 (experment autoimmune encephomeylophy,EAE)为其经典的动物模型。中枢神经系统轴突损伤后再生困难的主要原因是轴突损伤局部区域存在相关生长抑制分子[1],Rho激酶信号途径是介导上述相关抑制分子的重要转导通路。Rho相关激酶(Rho kinase,ROCK)属于丝/苏氨酸蛋白激酶,是Rho的下游靶效应分子。肌球蛋白轻链磷酸酶(myosin light chain,p-MLC)、LIM 激酶(LIM kinase 2,LIMK2)、脑衰反应调节蛋白(collapsin response mediator protein-2,CRMP-2)为ROCK较为重要的底物,ROCK被激活后可调节p-MLC、LIMK2与 CRMP-2的活性,使肌球蛋白磷酸酶磷酸化,进而使肌球蛋白磷酸化程度增高,影响肌动球蛋白系统而导致轴突生长锥的塌陷,抑制轴突生长[2]。Y-39983为ROCKⅡ的选择性抑制剂,本研究拟通过观察Rho/ROCK通路在EAE中的作用,探讨Y-39983对Rho/ROCK通路的影响及可能作用机制。
1 材料和方法
1.1 材料
1.1.1 实验动物及分组:雌性Lewis大鼠(清洁级)100只,体质量150~280g,由中山大学动物实验中心提供(粤监证号2005A061);豚鼠10只,体质量320~360g,由广州医学院动物实验中心提供,用于制作脑脊髓白质抗原。
1.1.2 主要药品与试剂:不完全福氏佐剂购自北京鼎国生物技术有限公司;吸附无细胞白百破联合疫苗购自武汉生物制品研究所;卡介苗购自上海生物制品研究所;Y-39983、一抗蛋白脂蛋白(PLP)、髓磷脂碱性蛋白(MBP)、3-环腺苷酸-3-磷酸二酯酶(CNPase)、p-MLC、CRMP-2及 LIMK2均购自美国Sigma公司。在不完全福氏佐剂中按10∶1比例加入卡介苗混合而成完全福氏佐剂(CFA)。麻醉处死豚鼠,按1g白质加1mL PBS的比例,在冰盒中研磨,制成牛乳样组织匀浆。将该匀浆与等体积CFA充分混匀制成诱导乳剂。
1.2 方法
1.2.1 动物模型的建立、治疗及平均临床评分:将大鼠随机分为对照组(健康大鼠)、EAE模型组(模型组)及干预组,每组均30只。模型组及干预组建立EAE动物模型,对照组为健康大鼠。
EAE模型制作过程:按0.12mL/100g比例给予模型组及干预组中的每只Lewis鼠双后足垫皮下注射诱导乳剂,于后足垫背面真皮内辅以注射0.2mL吸附无细胞白百破联合疫苗进行模型制作。致敏当天记为第0天[3]。对照组用生理盐水代替诱导乳剂。
于模型动物发病后第1天开始并连续3d给予干预组大鼠腹腔注射 Y-39983(20μg/d),模型组同样于该组动物发病第1天给予连续3d腹腔注射生理盐水(20μg/d)。每天称体质量,观察动物行为,并每组随机抽取6只Lewis大鼠进行临床症状评分。
临床症状评分分5级:正常或无任何神经缺损症状,计为0分;尾部肌张力消失,可见轻度步态笨拙,计为1分;尾部无力,双后肢肌张力低,计为2分;尾部无力,双后肢瘫痪,但给予刺激后可挪动,记3分;瘫痪累及前肢,伴尿便失禁,计为4分;濒死状态或死亡,计为5分。症状介于两标准级之间以±0.5分计。动物出现临床症状即可认为发病。造模成功的动物进入下一步干预实验。造模及干预过程中2只死亡和3只未发病大鼠均退出实验。不足大鼠按相同方法补足。
1.2.2 组织学观察:(1)取材:于干预结束后第5天,各组随机抽取6只大鼠取出脑组织放入4%(质量浓度)多聚甲醛中固定、石蜡包埋、切片。(2)采用HE染色、搔洛花青髓鞘染色及电镜观察(具体步骤参见说明书),在40倍显微镜视野下分别观察各组脑组织中炎性细胞浸润程度、髓鞘脱失现象及髓鞘的超微结构。
1.2.3 APC免疫荧光染色法:(1)取材:于干预结束后第5天,各组随机抽取6只大鼠采用10%(体积分数)水合氯醛麻醉大鼠后处死,取出脑组织放入4%(质量浓度)多聚甲醛中固定、石蜡包埋、切取厚度为2mm的脑组织切片。(2)每组中均选取包含侧脑室旁脑组织区域的20张切片放入漂洗盒,PBS换洗3次,然后加入过氧化物酶阻断剂室温下振荡孵育30min;PBS换洗3次,加入正常非特异性血清,室温下振荡孵育60min;加入一抗APC(1∶125),振荡后于4℃冰箱中孵育过夜;取出后PBS换洗5次;加入生物素标记的Ⅱ抗,室温下振荡孵育45min,PBS换洗3次;加入链霉素抗生物素-过氧化物酶溶液,室温下振荡孵育30min,PBS换洗3次;最后采用显微镜40倍视野下进行观察摄片。观察各组脑组织中髓鞘的脱失变化情况,APC表达愈多说明髓鞘脱失愈严重。
1.2.4 采用蛋白免疫印迹(Western blot)法对PLP、MBP及 CNPase、p-MLC、CRMP-2、LIMK2蛋白定量测定:各组于干预结束后第5天均随机抽取10只大鼠进行麻醉后处死并灌注,取大鼠脑组织按照凯基核蛋白和胞质蛋白提取试剂盒说明书分别提取胞质蛋白和胞核蛋白;BCA蛋白定量,配制胶液并灌胶;10%(质量浓度)SDS-PAGE电泳分离;转膜,染色并照相,经TBST洗涤脱色;封闭2h;TBST洗涤4次;分别加入一抗PLP、MBP及CNPase、CRMP-2(1∶500)、LIMK2(1∶500)及p-MLC(1∶1000)稀释液振荡孵育过夜;TBST洗涤洗膜;加HRP标记的二抗稀释液室温振荡孵育1.5h;TBST洗膜;用化学发光法显色;将所得谱带照相并经图像分析软件分析相应谱带密度值。所测密度值越高,说明各蛋白表达量越高。
1.3 统计学处理 采用SPSS13.0软件进行分析。计量资料均采用均数±标准差表示。组间比较采用单因素方差分析,率的比较采用卡方检验。以P<0.05表示差异有统计学意义。
2 结果
2.1 临床症状 模型组与干预组均于免疫后第7~10天开始发病及免疫后第17~20天出现复发。干预组临床症状明显改善,其发病高峰期临床评分(4.229±1.017)低于模型组(1.076±0.682)(P<0.05);模型组发病高峰期持续(5.3±1.46)d,干预组为(3.7±1.35)d,短于模型组(P<0.05)。干预组中共有11只出现临床症状复发,复发率为有37.5%(11/30),低于模型组复发率〔46.67%(14/30)〕(P<0.05)。
2.2 脑组织病理表现 HE染色显示模型组中可见明显的炎性细胞浸润现象及血管袖套形成,干预组中炎性细胞浸润现象明显减少,血管袖套现象消失;搔洛花青髓鞘染色显示模型组中髓鞘明显肿胀及断裂,干预组中髓鞘脱失现象减轻;电镜观察可见模型组病变散在分布,髓鞘呈松散的层状结构,有脱失及融合,明暗相间消失,干预组上述改变均明显减轻(图1)。APC免疫荧光染色显示模型组中APC表达增多,干预组中APC表达较模型组减少(图2)。对照组上述方法均未发现明显异常改变。

图1 各组脑组织病理表现(×40)

图2 各组脑组织APC免疫荧光染色比较(×40)
2.3 髓鞘相关蛋白(PLP、MBP 及 CNPase)及ROCKⅡ底物(p-MLC、CRMP-2及 LIMK2)蛋白含量测定 PLP、MBP及CNPase含量模型组低于对照组,干预组高于模型组(P<0.05)。p-MLC、CRMP-2及LIMK2含量模型组明显高于对照组,干预组低于模型组(P<0.05)(表1,图3)。

表1 各组PLP、MBP、CNPase、CRMP-2、p-MLC及LIMK2密度值比较
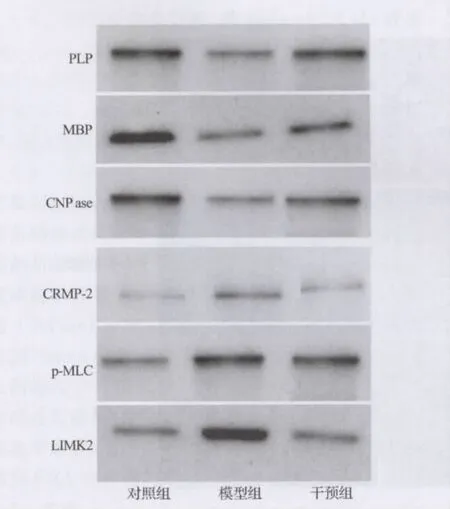
图3 各组脑组织中髓鞘相关蛋白(PLP、MBP及CNPase)及ROCKⅡ底物(p-MLC、CRMP-2及LIMK2)表达(Western blot)
3 讨论
中枢及外周有髓神经纤维的髓鞘部分或全部损害及轴突受损是EAE的典型病理特征[4]。本研究中组织病理学观察发现,EAE模型组动物脑组织轴突损害较健康对照组明显,并伴有较多炎性细胞浸润。
Rho相关激酶分为ROCKⅠ和ROCKⅡ型,其中ROCKⅡ在脑组织中表达明显,活化的ROCKⅡ对p-MLC的肌动蛋白结合亚基(MBS)肽链的Thr、Ser及Thr855进行磷酸化修饰,使p-MLC失活[5]。ROCKⅡ可使 CRMP-2磷酸化,从而阻碍肌动蛋白聚合和解聚过程,最终导致生长锥塌陷、回缩,轴突生长停止[6]。ROCKⅡ也可能通过LIMK2抑制coffilin介导的肌动蛋白解聚和其他一些调节蛋白,如肌动蛋白解聚因子(ADF)活性作用于肌动-球蛋白系统,从而阻碍肌动蛋白聚合和解聚过程,最终导致生长锥塌陷、回缩,轴突生长停止[7]。本研究结果显示,EAE模型组中的MLCP、CRMP-2及LIMK2含量较健康对照组明显升高,说明在EAE的发病过程中,ROCK的激活可通过调节上述激酶底物含量从而抑制神经轴突的生长,提示Rho/ROCK通路参与了EAE的发生。
Y-39983是ROCKⅡ选择性抑制剂,其可通过与Rho激酶催化结构ATP位点结合而抑制ROCKⅡ激活,同时还可抑制炎性因子,如肿瘤坏死因子的增殖[8]。本研究采用Y-39983进行干预后发现,Y-39983可以降低EAE模型的发病率、减轻临床症状及炎性细胞浸润程度和髓鞘脱失。同时,通过蛋白印迹方法检测结果显示Y-39983干预组中 ROCK 底物蛋白 pMLC、CRMP-2 及LIMK2含量较EAE模型组明显降低,主要髓鞘相关蛋白PLP、MBP及CNPase含量较EAE模型组显著增高,由此可以推测Y-39983可能通过抑制ROCKⅡ激活,使Rho激酶下游通路部分中断,减少破坏轴突生长的底物蛋白生成,促进髓鞘形成及轴突再生的主要作用机制之一。
[1]尚宏,付锦,王鹏军,等.一氧化氮供体对实验性自身免疫性脑脊髓炎的作用[J].中国神经免疫学和神经病学杂志,2011,18(2):79-82.
[2]Keller T,Yamaguchi A,Iwata N,et al.The therapeutic effects of Rho-ROCK inhibitors on CNS disorders[J].Clin Risk Manag,2008,4(3):605-615.
[3]焦卓敏,富羽弘,王维治.实验性自身免疫性脑脊髓炎动物模型的比较[J].中国神经免疫学和神经病学杂志,2005,12(5):258-261.
[4]Lassmann H.Experimental models of multiple sclerosis[J].Rev Neurol,2007,163(6):651-655.
[5]Nahm M,Long AA,Pail SK,et al.The cdc42selective GAP rich regulates postsynaptic development and retrograde BMP transsynaptic signaling[J].Cell Biol,2010,191(3):661-675.
[6]Zhang G,Lehmann HC,Manoharam S,et al.Anti-ganglioside antibody mediated activation of RhoA induces inhibition of neurite outgrowth[J].Neurosci,2011,31(5):1664-1675.
[7]Therapeutic potential of experimental autoimmune encephalomyelitis by Fasudil,a Rho-kinase inhibitor[J].Neurosci,2010,13(1):86-94.
[8]Tokushige H,Inatani M,Nemoto S,et al.Effects of topical administration of Y-39983,a selective rho-associated protein kinase inhibitor,on ocular in rabbits and monkeys[J].Invest Ophthalmol,2007,48(1):3216-3222.
猜你喜欢
中华耳科学杂志(2022年1期)2022-11-24 15:09:22
生物化学与生物物理进展(2022年11期)2022-03-02 11:34:48
浙江临床医学(2021年12期)2021-11-29 14:43:18
天津医科大学学报(2021年3期)2021-07-21 09:03:46
世界科学技术-中医药现代化(2021年12期)2021-04-19 12:31:40
党的生活(黑龙江)(2018年9期)2018-10-17 01:24:24
益寿宝典(2018年1期)2018-01-27 01:50:24
中国医药生物技术(2015年4期)2015-12-26 08:26:36
吉林大学学报(医学版)(2015年1期)2015-12-17 07:47:27
中华神经创伤外科电子杂志(2015年1期)2015-01-21 09:09:24
