
磷钨酸“瓶中船”型催化剂的合成、表征及催化分解NO x性能
2013-09-15 01:41张学杨
无机化学学报 2013年2期
张学杨 王 睿
(山东大学环境科学与工程学院,济南 250100)
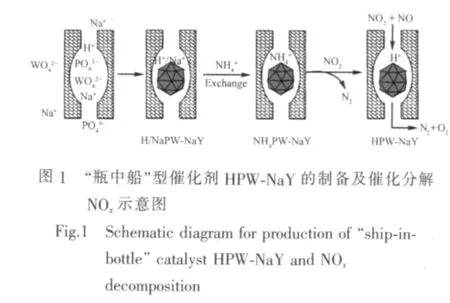
杂多酸因其优良的氧化还原性及强酸性而被广泛应用于催化领域[1-7],然而其本身的低热稳定性、低比表面积以及在液相中高的溶解度制约了它在多相催化中的工业应用。为克服上述缺陷,引入各种载体材料所制备的负载型杂多酸,在增大了比表面积的同时也提高了其热稳定性[8-12],然而杂多酸从载体材料上的流失仍不可避免。Sulikowski等[13]提出的将杂多酸固载于沸石超笼中形成 “瓶中船”(Ship-in-Bottle)型催化体系的构想,解决了杂多酸的流失问题,在随后的研究中还发现该类型催化剂较单纯的杂多酸在多相催化反应中具有更高的催化活性[13-16]。为提高“瓶中船”型催化剂的制备效率,人们开展了诸多研究[17-20],其中Hou等[20]引入的微波辐射技术提高催化剂制备效率的效果最为显著,然而由于在催化剂制备过程中Na+(原料中的NaH2PO4及Na2WO3)的存在,沸石笼中的生成物除杂多酸外还有杂多酸钠盐,目前仍无法制备只含有单纯杂多酸的“瓶中船”型催化剂。
Moffat等[21-23]的研究表明,磷钨酸铵(NH4PW)可与NO2发生氧化还原反应12NO2+4(NH4)3PW12O40→12N2+3O2+18H2O+4H3PW12O40,随着反应的进行,还原剂NH4PW逐渐转化为磷钨酸(HPW)。我们由此得到启发,先将沸石笼中的磷钨酸及其钠盐通过复分解反应全部转化为NH4PW,而后通过NO2与NH4PW的反应将沸石笼中的NH4PW全部转化为HPW,从而制备出沸石笼中只含有HPW的 “瓶中船”型催化剂。
如图1所示,在微波辅助条件下,于NaY沸石超笼中原位合成磷钨杂多酸及其钠盐,而后通过取代Na+与H+的方式制备出“瓶中船”型催化剂NH4PW-NaY,进而通过氮氧化物(NOx)与该催化剂反应制备出沸石超笼中只含有HPW的“瓶中船”型催化剂HPW-NaY。随后考察了该催化剂对NOx的吸附、脱附及催化分解性能。
1 实验部分
1.1 催化剂制备
NH4PW-NaY“瓶中船”型催化剂的制备:参照文献[20],首先将 NaY 沸石(nSi∶nAl=30∶1)于 550 ℃马弗炉中煅烧3 h以去除沸石笼中的水。称取1.074 g磷酸氢二钠与11.875 g钨酸钠混合于50 mL烧杯,加20 mL蒸馏水后将烧杯移至70℃水浴锅,磁力搅拌下使其溶解,称取3.0 g煅烧后的NaY沸石,在剧烈搅拌下加至上述混合液。继续搅拌20 min后逐滴加入浓盐酸调节混合液pH值至1.0,并将烧杯移入MCL-2型微波化学实验仪(四川大学无线电系)中,于700 W功率下照射7 min。过滤烧杯中混合液,并用80℃蒸馏水洗涤滤出物10次,以除去残留于NaY沸石外的杂多化合物,将滤出物置于干燥箱中,105℃干燥过夜,200℃下煅烧3 h,得到NaY沸石超笼中含有磷钨酸及其钠盐的“瓶中船”型样品(标注为H/NaPW-NaY),研磨备用。称取1.441 g氯化铵于50 mL烧杯,加20 mL蒸馏水使其溶解,将上述制备的H/NaPW-NaY在剧烈搅拌下加入氯化铵水溶液,将烧杯移入KQ-50型超声反应器(昆山市超声仪器有限公司),在60℃下超声处理20 min。处理完毕后过滤混合液,将滤出物用80℃蒸馏水洗涤3次除去氯化钠及过量氯化铵,置于干燥箱中,105℃干燥过夜,即得NaY沸石超笼中只含有NH4PW的“瓶中船”型催化剂(标注为NH4PW-NaY),备用。
NH4PW 的制备:按化学计量比(nNH4Cl∶nHPW=3∶1)称取NH4Cl与HPW分别配成溶液,在剧烈搅拌下,将NH4Cl逐滴加入HPW水溶液,产生白色沉淀,过滤,干燥,得NH4PW,备用。
1.2 催化剂活性评价
NOx吸附脱附实验装置如图2所示。
吸附过程:采用质量流量计精确控制NO、O2和N2流量,调节混合气中NO浓度为1339 mg·m-3,O2为8vol%,蒸汽发生器产生4.5vol%水蒸汽,N2为平衡气,三路气体于缓冲瓶中混合均匀,吸附试验在内径为8 mm的石英管反应器中进行,取1.0 g催化剂置于反应器中间,两端以石英棉固定,吸附温度为170℃,吸附过程中NOx浓度由TH-9905型NO/NO2分析仪(武汉市天虹仪表有限责任公司)测定。
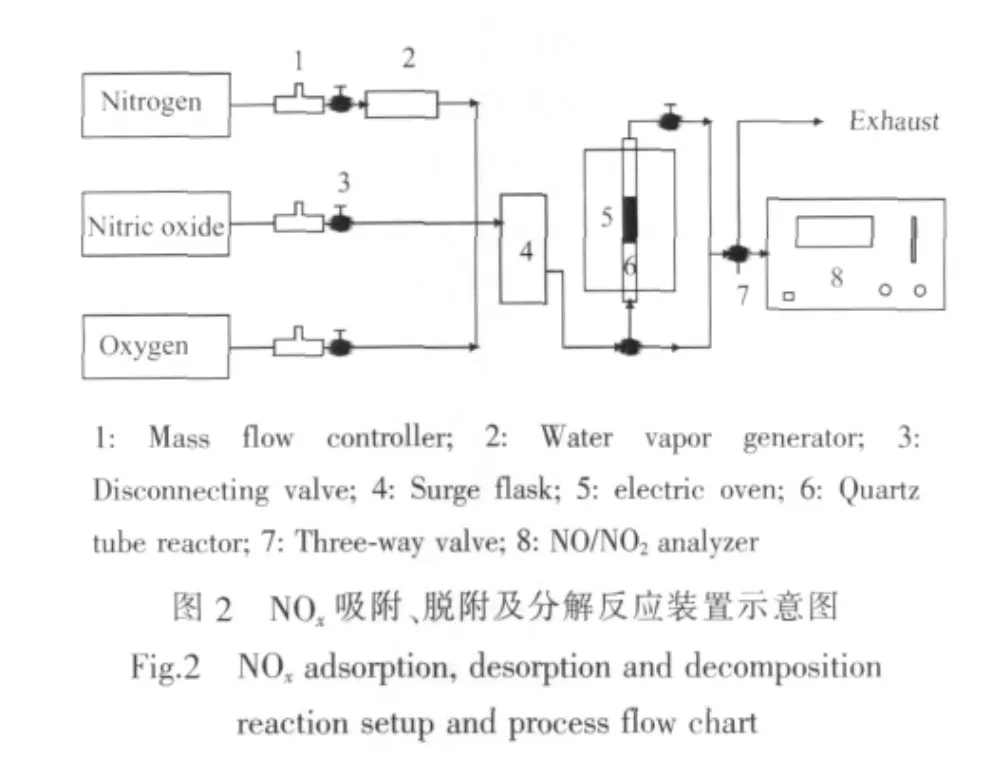
脱附过程:吸附NOx完毕后关闭气路,采用降温通水蒸汽方式脱附NOx,对饱和吸附NOx的催化剂,关闭电阻炉使其自170℃降至室温,降温伊始调节蒸汽发生器,向气路中通10vol%水蒸汽以脱除催化剂上所吸附的NOx,脱附过程中NOx浓度由NO/NO2分析仪测定。
分解过程:采用LZL-204型质谱仪(北京分析仪器厂)进行程序升温脱附-质谱(TPD-MS)测试,用于NOx催化分解及分解产物的检测。吸附NOx后的样品约300 mg置于程序升温装置的石英管反应器中,以20 mL·min-1的高纯氦气吹扫气路1 h后,以预定的升温速率自30℃程序升温至500℃,分解过程中以He为载气,分解产物通过质谱检测,根据质核比(m/z)进行离子流选择,m/z=28为N2,30为NO,32 为 O2,44 为 N2O,46 为 NO2。在质谱分析中离子碎片的影响不可忽略,在质谱检测的结果分析中对离子碎片的影响进行修正。
1.3 催化剂表征
傅立叶变换红外光谱 (FTIR)使用美国Nicolet Instrument公司产Nicolet 5DXC红外光谱仪进行分析。样品使用KBr压片,扫描范围为4 000~400 cm-1,分辨率为 2 cm-1。
X射线粉末衍射(XRD)分析采用德国Bruker公司的Bruker D8 Advanced型粉末衍射仪进行。射线源采用波长为0.154 05 nm的Cu Kα线,石墨单色器,管压40 kV,管电流60 mA,Ni滤波片,扫描范围 10°~45°,扫描速率 0.02°·s-1。
吡啶吸附红外(Py-IR)分析,将样品置于红外池,在压力为0.1 Pa,温度为200℃条件下脱气4 h后,进行吡啶吸附,吸附后采用美国Nicolet Instrument公司生产的Nikolet 560红外光谱仪进行分析。
催化剂的比表面积及孔容采用北京贝士德仪器科技有限公司的3H-2000PS2型静态容量法比表面及孔径分析仪测定。样品于150℃真空状态下加热除气2 h后,在液氮温度-196℃下获得样品的N2吸附-脱附等温线。
2 结果与讨论
2.1 催化剂NH 4PW-NaY的表征
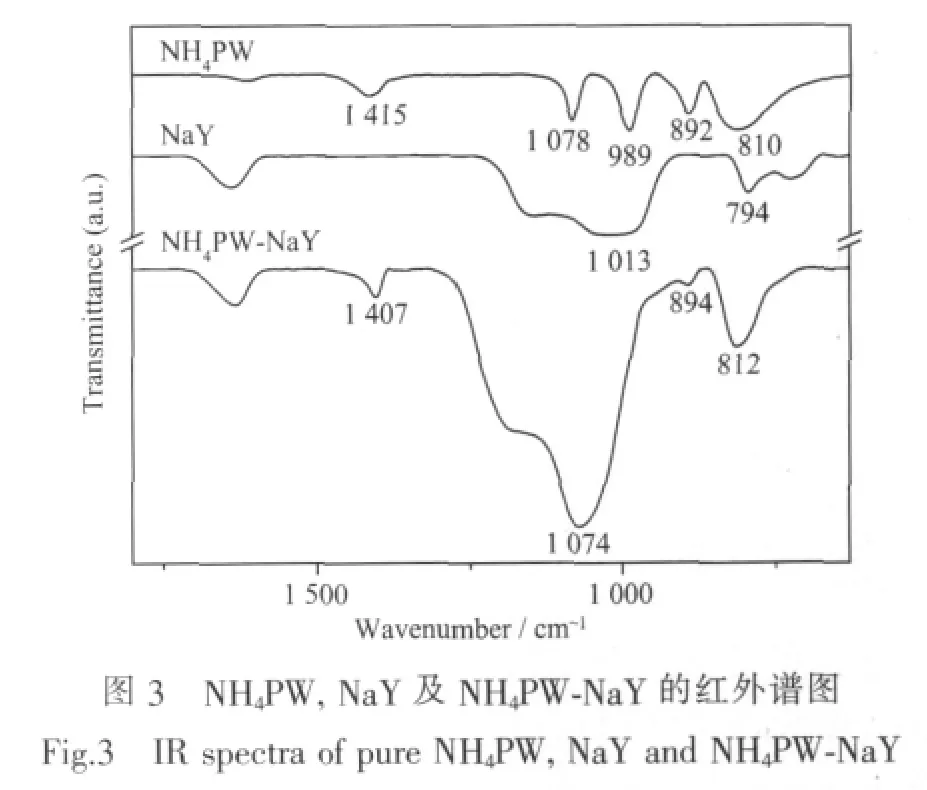
所制备的 NH4PW、NH4PW-NaY及 NaY的红外光谱如图3所示。样品NH4PW的红外光谱中有Keggin结构杂多阴离子的4个特征峰[6],即位于1 078 cm-1的P-O振动峰,位于892 cm-1的W-Ob-W振动峰,位于810 cm-1的W-Oc-W振动峰,以及位于989 cm-1的W=O振动峰。微波法所制备的样品NH4PW-NaY,位于989 cm-1的W=O振动峰被NaY沸石中强烈的Si-O键的振动峰所遮蔽,而其他3个Keggin结构特征峰P-O、W-Ob-W及W-Oc-W分别在1 074、894及812 cm-1处出现,以上Keggin结构特征峰以及在1 407 cm-1处出现的NH4+振动峰均表明NH4PW在NaY沸石超笼内被原位合成。
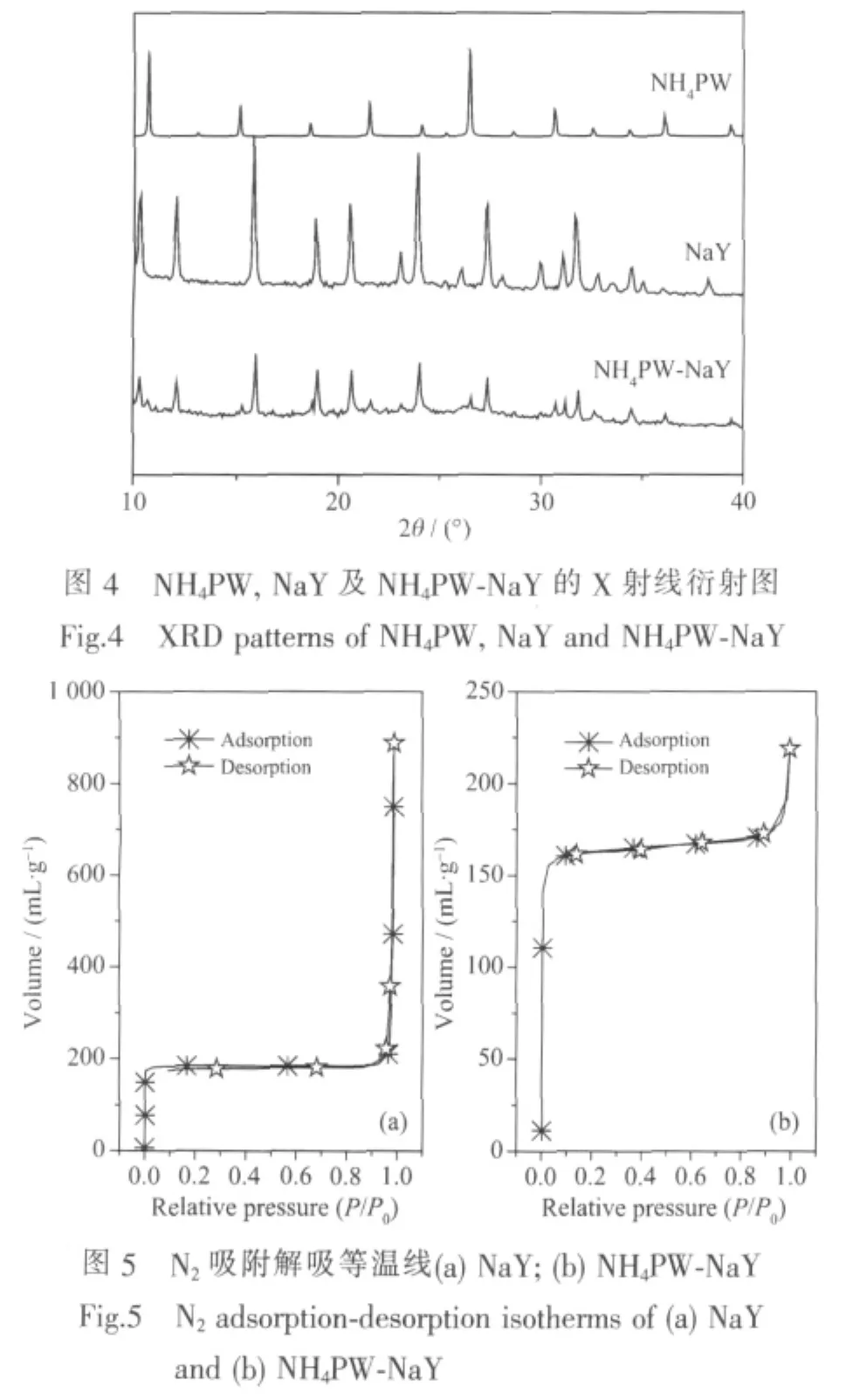
NH4PW、NaY及NH4PW-NaY的X射线衍射图如图4所示。采用微波辐射法制备的催化剂保留了NaY沸石笼的结构,且NH4PW-NaY样品中无明显NH4PW的XRD晶形,由此说明沸石超笼外无NH4PW存在,结合FTIR结果可知,所合成的NH4PW位于沸石超笼内,这一结果与Hou[20]以USY沸石所制备的“瓶中船”的表征结果吻合。
N2在NaY及NH4PW-NaY上的吸附-脱附等温线如图5所示,该两种样品均表现出典型的Ⅰ型等温线,表明两者均为微孔材料。NaY及NH4PW-NaY的形貌结构数据如表1所示,NaY沸石的比表面积为693 m2·g-1,其沸石超笼中原位合成NH4PW后催化剂NH4PW-NaY的比表面积则降为591 m2·g-1,原位合成NH4PW后NaY沸石笼的孔体积则由1.2 mL·g-1下降至 0.1 mL·g-1,这主要是由于 NH4PW 原位生成于NaY沸石笼后,填充了沸石笼的空间,因而造成了比表面积及孔容积的下降。对NaY及NH4PW-NaY的形貌分析进一步证实了NH4PW被成功合生成于NaY沸石超笼中形成“瓶中船”型催化剂。

表1 NaY及NH4PW-NaY比表面积及孔容Table 1 Specific surface area and pore volume of NaY and NH 4PW-NaY
2.2 催化剂HPW-NaY的形成
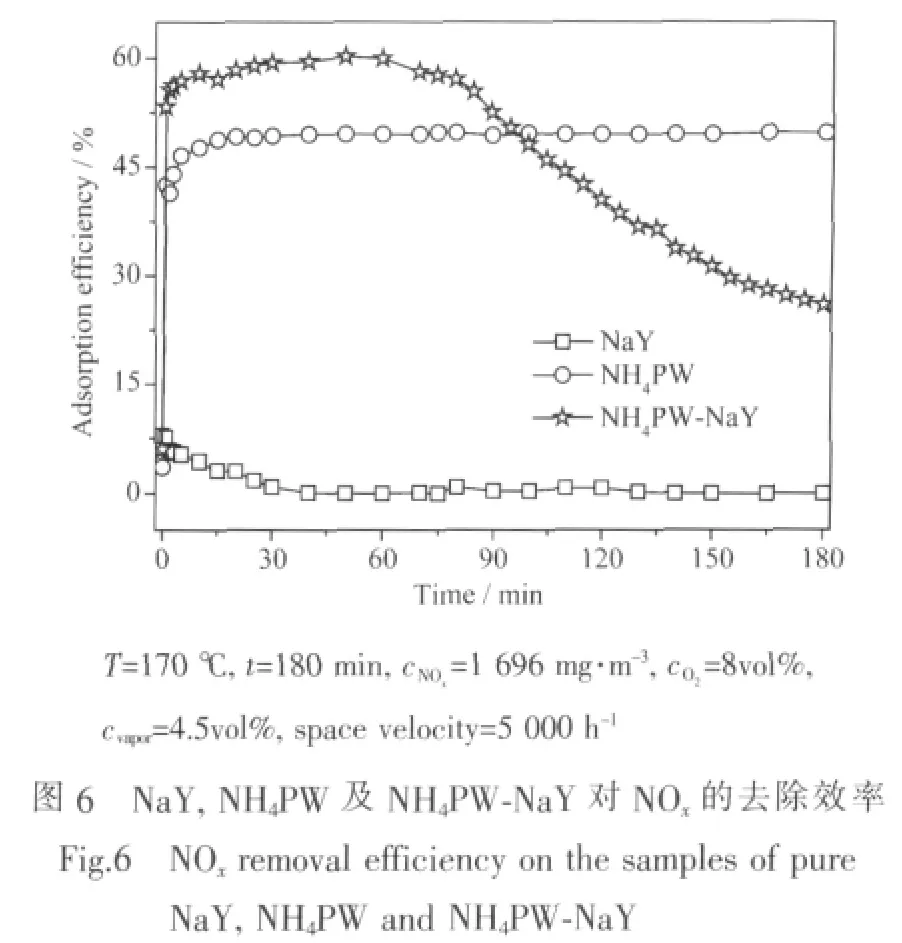
Moffat等[22]在研究NH4PW去除NO2的过程中发现,NH4PW可以与NO2反应生成HPW。为将NaY沸石笼中的NH4PW全部转化为HPW得到HPWNaY催化剂,以NOx为氧化剂与NH4PW-NaY进行充分反应。图6显示了NH4PW-NaY、NH4PW及NaY对NOx的去除效率,以上3种样品对NOx的去除研究均为在170℃下进行,且NOx的初始浓度均为1 696 mg·m-3。结果表明,NaY 沸石对 NOx的去除率低于 8%,NH4PW 对 NOx的去除率可达 50%,而NH4PW-NaY对NOx的去除效率最高为60%。NH4PW-NaY之所以较NaY及NH4PW有着更高的NOx去除效率,主要是由于NH4PW在NaY上高度均匀分散,从而提高了NH4PW的比表面积,进而增大了NH4PW与NOx的接触几率所致。由于NH4PW在NaY沸石中含量较少,因此反应至80 min时随着NaY沸石中NH4PW的不断消耗,NH4PW-NaY对NOx的去除率逐渐降低,反应至100 min时NH4PWNaY对NOx的去除率已低于单纯的NH4PW。
当NaY沸石笼中NH4PW与NOx反应完全后,NH4PW-NaY转化为HPW-NaY,对反应前后的NH4PW-NaY进行FTIR表征以证实HPW-NaY的形成,结果如图7所示。NH4PW-NaY与NOx反应后,磷钨杂多阴离子的Keggin特征峰未发生改变,而在1 407 cm-1位置的吸收峰消失,且在1 384 cm-1处出现新的吸收峰。吸收峰的消失表明原催化剂中的NH4+与NOx反应完全,即NH4PW均已转变为HPW。而1 384 cm-1位置的吸收峰被指认为吸附于HPW上的NOx在进行红外压片时与KBr产生的结合键[24-25],该吸附峰的出现表明了NOx在HPW上有大量的吸附,由此进一步表明了HPW的形成。NH4PW-NaY与NOx反应前后的FTIR结果表明NH4PW-NaY已转化为HPW-NaY,且有大量的NOx吸附于新生成的HPW-NaY。
为进一步验证“瓶中船”型催化剂HPW-NaY的形成,对NH4PW-NaY及HPW-NaY进行吡啶吸附红外研究,结果如图8所示。在1 600 cm-1与1 450 cm-1附近的红外吸附峰被指认为Lewis型吡啶吸附峰,1 540 cm-1处的红外峰被指认为Brønsted型吡啶吸附峰,而在1 490 cm-1处的吸附峰则为Lewis型与 Brønsted 型吡啶吸附峰之和[20]。Brønsted酸与Lewis酸相对强度可以通过处于1 540与1 450 cm-1位置的红外峰进行计算,计算公式为[26]:酸型吡啶红外吸附比;εL/εB是 Brønsted 型与 Lewis型吡啶红外吸附消光系数比,Stencel等[27]研究表明,对于nSi/nAl比高于15的沸石,该值可取为1.5。对NH4PW-NaY及HPW-NaY的吡啶吸附红外结果进行分析,结果示于表2。催化剂NH4PW-NaY中Brønsted 酸与 Lewis酸强度比 AB/AL为 0.336,与NOx反应并转化为HPW-NaY后,该比值升高为0.440,这表明反应后Brønsted酸位显著增加,而该酸位的增加是由于NH4+转化为而H+所致。因此,吡啶吸附红外的结果进一步证实了NH4PW-NaY与NOx反应产物为HPW-NaY,沸石超笼中只含有HPW的“瓶中船”型催化剂HPW-NaY被成功制备。
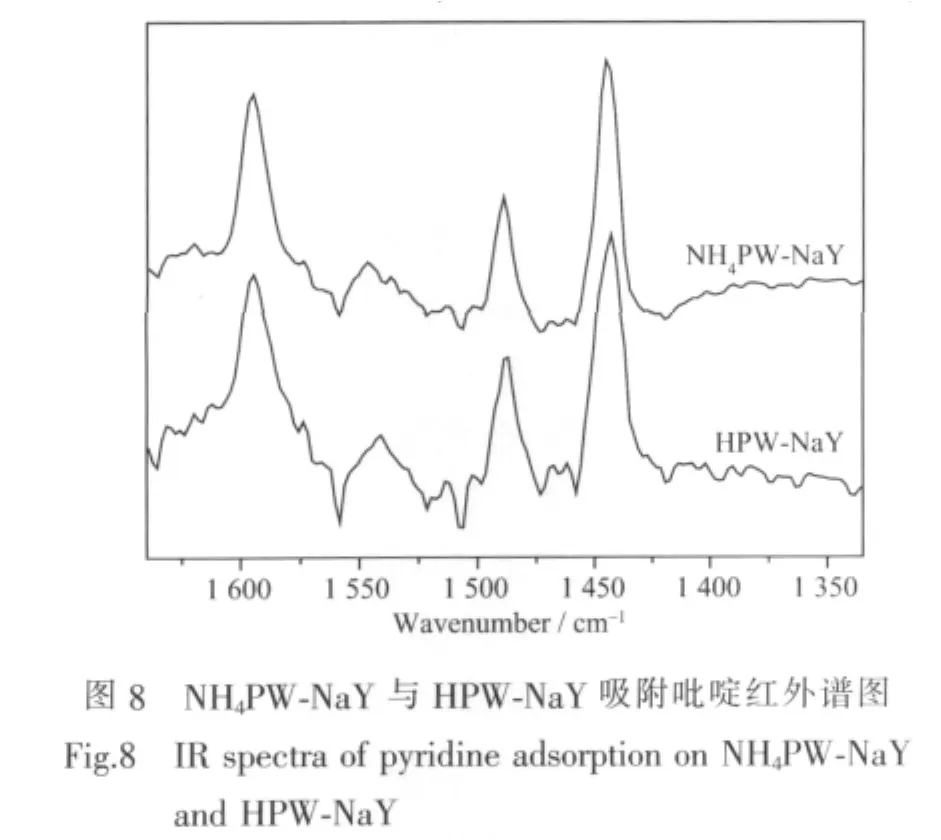

表2 NH 4PW-NaY与HPW-NaY的酸强度比较Table 2 Acidity comparison between NH 4PW-NaY and HPW-NaY
2.3 NO x在HPW-NaY上的吸附与脱附
HPW具有在较低温度下吸附NOx的能力[12,24,28-32],且众多研究表明[25,33-34],在HPW吸附NOx过程中,NOx与极性分子H2O竞争吸附于HPW上,在50°C以下时H2O较NOx具有吸附优势,易于吸附于HPW的二结结构中形成带有6个结晶水的磷钨酸H3PW12O40·6H2O,而150℃时NOx则表现出较H2O更强的吸附优势,每一个NOx取代HPW中2个结晶水形成H3PW12O40·3NOx。由此可见,吸附有NOx的杂多酸,除了可以通过提高温度使NOx发生热脱附外,还可以在降低温度的条件下借助通入的水蒸汽使NOx被H2O反取代的方式进行脱附,该方法可以避免传统升温热脱附中温度过高对HPW结构带来的破坏。
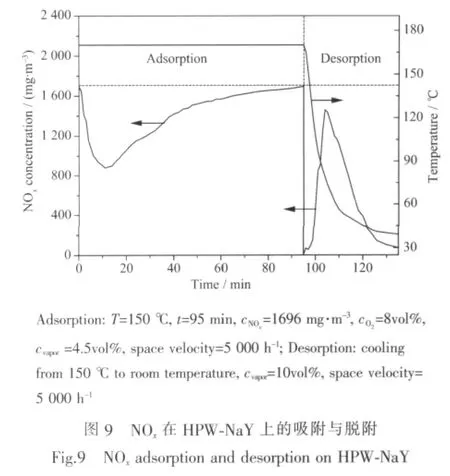
图9示出了NOx在HPW-NaY上的吸附及降温通水蒸汽方式进行的脱附效果。在170℃条件下,HPW-NaY对 1 696 mg·m-3的 NOx进行了吸附,通过对该吸附曲线进行积分得到HPW-NaY对NOx的吸附容量为 2.38 mgNOx·gcat-1。饱和吸附NOx后对HPW-NaY进行降温,通入水蒸汽脱附NOx,结果发现当温度下降至100℃时,吸附于HPW-NaY的NOx发生了脱附,对脱附曲线进行积分计算,发现降温通水蒸气脱附法可使83%的NOx在40 min内被脱附。低温通水蒸汽的脱附方式可避免杂多酸在高温下发生分解,此外水蒸汽及时补充了NOx脱除后在催化剂上所形成的空位,因此在NOx脱附后HPW-NaY的结构未受到破坏并迅速恢复至吸附NOx前的初始状态,该催化剂无需再生即可进行重复使用。对该催化剂进行重复使用,再次吸附NOx后进行随后的TPD-MS实验。
2.4 NO x在HPW-NaY上的催化分解
将降温通水蒸汽脱附NOx后的催化剂HPWNaY再次饱和吸附NOx,而后采用TPD-MS进行HPW-NaY对NOx的催化分解研究,结果如图10a所示,为进行比较,HPW对NOx催化分解的TPDMS结果列于图10b。
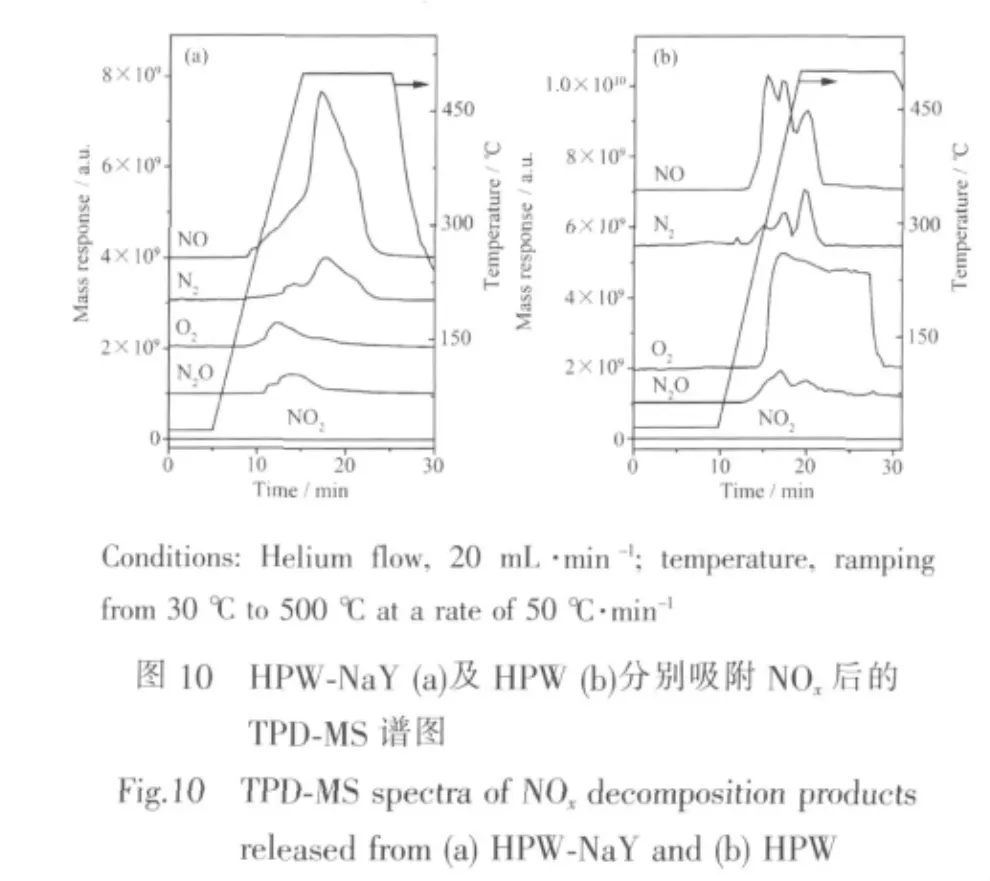
以HPW-NaY及HPW为催化剂催化分解NOx过程中,分解产物为NO、N2O、N2及O2,且均未检测到NO2,这一结果与McCormick等[35]以HPW为催化剂进行NOx催化分解的结果相似。在NOx催化分解过程中,O2的脱附是最为关键的一步,在此前的关于杂多酸催化剂催化分解NOx报道中,由于分解在有O2的存在下进行[24,35],因此未讨论分解过程中氧的产生及脱附,本文作者[36]曾采用TPD-MS研究HPW及负载于多壁碳纳米管的HPW催化分解NOx中首次发现了O2的产生,证实了分解所产生的氧原子可以由杂多酸催化剂上脱附。载体材料在NOx催化分解中直接影响到氧原子的脱附,从而影响NOx的催化效率,该实验首次探讨了以NaY沸石为载体的催化剂HPW-NaY催化分解NOx中氧的产生及脱附。
如图10所示,与N2O、N2及O2相比,NO在最低的分解温度下开始出现,这是由于催化剂吸附NOx过程中,部分NOx以物理吸附形式吸附于催化剂中,物理吸附的NOx与催化剂的结合能较弱,因此在较低的热作用下,NOx即发生脱附及分解。根据文献[37]的研究,NOx分解中产物N2O是由NO的歧化反应而来。分解产物中的N2则主要来自于NO,由于HPW的催化作用使N-O键发生了断裂,产生活性氮原子和氧原子,其中,活性氮原子相互碰撞形成N2分子而被释放。然而产生的氧原子则较为活泼,易于与杂多酸催化剂中外层金属原子W相接合形成臭氧型加合物[38]。由于端氧Od位较其他位置氧有更高的反应活性[6],因此端氧位为最可能的接合氧原子位。该过氧杂多化合物为反应中间产物,其氧原子易于脱出并相互结合为O2进而释放,由于氧原子在相互结合为O2并释放出之前经历了加合与脱离这一复杂过程,因而其产生要晚于N2。
催化剂HPW及HPW-NaY催化分解NOx的转化率、N2及N2O的选择性如表3所示。HPW-NaY催化分解NOx的转化率为61%,其中N2选择性为75%,而N2O选择性仅为25%。催化剂HPW对NOx的转化率为54%,N2及N2O的选择性分别为53%与47%,由此可见,催化剂HPW-NaY在NOx催化分解中性能优于单纯的HPW催化剂。由于HPW限域于NaY沸石笼中,因此HPW催化剂所吸附的NOx分子同样处于沸石笼中,在快速升温分解过程中,部分NOx会由HPW催化剂中脱附,由于NOx分子受沸石笼的约束,所脱附的NOx仍处于HPW催化剂表面,因此催化剂对NOx仍有催化分解作用。而单纯的HPW催化剂上升温所脱附的NOx很快脱离HPW固体表面而未被催化分解,仅有升温过程中残留于HPW上的少量NOx及脱附后未及时脱离HPW表面的NOx参与了催化分解,由此可见,“瓶中船”型催化剂HPW-NaY对NOx的催化活性高于HPW。

表3 NO x(x=1,2)在催化剂HPW及HPW-NaY上转化率及N2、N2O选择性Table 3 Conversion of NO x and selectivities to N2 and N2O with HPW and HPW-NaY
3 结 论
(1)FTIR、XRD、比表面积及孔体积测试结果表明,微波辐射条件下可将12-磷钨杂多化合物原位合成于NaY沸石超笼中,制备出 “瓶中船”型NH4PW-NaY。
(2)NH4PW-NaY与NOx反应制备HPW-NaY的过程中,由于NH4PW-NaY的有效还原成分NH4PW高度分散于NaY沸石超笼中,因此其在反应温度为170℃时,对1 696 mg·m-3的NOx具有高达60%的去除效率,高于同条件下单纯的NaY与NH4PW对NOx的去除率之和。随着反应的进行,经FTIR及红外吸附吡啶测试表明NaY沸石笼中的NH4PW转化为HPW,由此沸石笼中只含有HPW的“瓶中船”型催化剂HPW-NaY被首次成功制备。
(3)HPW-NaY对NOx的吸附-脱附实验表明,在170℃HPW-NaY对NOx的吸附容量为2.4 mgNOx·g-1,对饱和吸附NOx的HPW-NaY采用降温通水蒸汽的方式可使83%的NOx在40 min内脱附,脱附的起始温度为100℃,且脱附后的催化剂可重复使用。
(4)对吸附NOx的HPW-NaY进行TPD-MS测试,研究其对NOx的催化分解性能,试验首次发现NOx在以NaY沸石为载体的HPW上的分解过程中有O2产生,且滞后于N2O及N2,HPW-NaY催化分解NOx的转化率及N2选择性分别为61%与75%,均高于单纯HPW催化剂。
[1]WANG En-Bo(王恩波),HU Chang-Wen(胡长文),XU Lin(许林).Introduction to Polyoxometalate Chemistry(多酸化学导论).Beijing:Chemical Industry Press,1998.
[2]LIU Ding(刘丁),TAN Hua-Qiao(谭华桥),CHEN Wei-Lin(陈维林),et al.Chinese J.Inorg Chem.(Wuji Huaxue Xuebao),2011,27:491-496
[3]ZHANG PENG(张鹏),WANG Een-Bo(王恩波),XING Chang-Yu(刑长宇),et al.Chinese J.Inorg.Chem.(Wuji Huaxue Xuebao),2009,25:560-562
[4]ZHANG Jin(张进),TANG Ying(唐英),LUOXi(罗茜),et al.Chinese J.Inorg.Chem.(Wuji Huaxue Xuebao),2004,20:935-940
[5]Li G,Ding Y,Wang J,et al.J.Mol.Catal.A:Chem.,2007,262:67-76
[6]Kozhevnikov I V.Catalysis by Polyoxometalates:Catalysis for Fine Chemical Synthesis,UK:John Wiley&Sons Ltd,2002.
[7]Wu Q,Hao X,Feng X,et al.Inorg.Chem.Commun.,2012,22:137-140
[8]WANG Bang-Jin(王帮进),DUAN Ai-Hong(段爱红),HU Jin-Feng(忽锦峰).J.Yunan Norm.Univ.:Nat.Sci.Edn.(Yunnan Shifan Daxue Xuebao:Ziran Kexue Ban),2010,30:40-42
[9]Bhorodwaj S K,Dutta D K.Appl.Catal.A,2010,378:221-226
[10]Gagea B C,Lorgouilloux Y,Altintas Y,et al.J.Catal.,2009,265:99-108
[11]GONG Da-Chun(龚大春),ZENG Fang(曾芳),GONG Dong-Ping(龚东平),et al.J.Chin.Three Gorges Univ.:Nat.Sci.Edn.(Sanxia Daxue Xuebao:Ziran Kexueban),2007,29:269-271
[12]Gómez-García M A,Pitchon V,Kiennemann A.Catal.Today,2005,107-108:60-67
[13]Sulikowski B,Haber J,Kubacka A,et al.Catal.Lett.,1996,39:27-31
[14]Mukai SR,Masuda T,Ogino I,et al.Appl.Catal.A,1997,165:219-226
[15]Mukai SR,Ogino I,Lin L,et al.React.Kinet.Catal.Lett.,2000,69:253-258
[16]GUOMai-Ping(郭麦平),WANGJun(王军),ZHOU Hua-Lan(周华兰),et al.Acta Chim.Sinica(Huaxue Xuebao),2008,66:1159-1162
[17]Pamin K,Kubacka A,Olejniczak Z,et al.Appl.Catal.A,2000,194-195:137-146
[18]Mukai SR,Lin L,Masuda T,et al.Chem.Eng.Sci.,2001,56:799-804
[19]Mukai SR,Shimoda M,Lin L,et al.Appl.Catal.A,2003,256:107-113
[20]Jin D,Gao J,Hou Z,et al.Appl.Catal.A,2009,352:259-264[21]Bélanger R,Moffat JB.J.Mol.Catal.A:Chem.,1996,114:319-329
[22]Blanger R,Moffat JB.Appl.Catal.B,1997,13:167-173
[23]Blanger R,Moffat JB.Catal.Today,1998,40:297-306
[24]Yang R T,Chen N.Ind.Eng.Chem.Res.,1994,33:825-831
[25]Chen N,Yang R T.J.Catal.,1995,157:76-86
[26]Borade A,Sayari A,Adnot A,et al.J.Phys.Chem.,1990,94:5989-5994
[27]Rhee K H,Udaya V,Rao S,et al.Zeolites,1983,3:337-343
[28]Belanger R,Moffat JB.J.Catal.,1995,152:179-188
[29]Hamad H,Soulard M,Lebeau B,et al.J.Mol.Catal.A:Chem.,2007,278:53-63
[30]Labaki M,Mokhtari M,Brilhac J F,et al.Appl.Catal.B,2007,76:386-394
[31]Landau M,Rao P,Thomas S,et al.Top.Catal.,2007,42-43:203-207
[32]MA Tao(马 涛 )WANG Rui(王 睿 ).Prog.Chem.(Huaxue Jinzhan),2008,20:798-810
[33]Hodjati S,Petit C,Pitchon V,et al.J.Catal.,2001,197:324-334
[34]Micek-Ilnicka A.J.Mol.Catal.A:Chem.,2009,308:1-14
[35]McCormick R L,Boonrueng S K,Herring A M.Catal.Today,1998,42:145-157
[36]ZHANG Xue-Yang(张学杨),CHENG Lin(程琳),YANG Feng(杨烽),et al.Chem.J.Chin.Univ.(Gaodeng Xuexiao Huaxue Xuebao),2012,33:1818-1826
[37]Li Y,Armor JN.Appl.Catal.,1991,76:L1-L8
[38]Hiskia A,Papaconstantinou E.Inorg.Chem.,1992,31:163-167
猜你喜欢
煤气与热力(2021年9期)2021-11-06
湖南饲料(2021年3期)2021-07-28
化工学报(2020年4期)2020-05-28
今日农业(2019年11期)2019-08-13
天然气化工—C1化学与化工(2019年6期)2019-02-18
物理教师(2018年5期)2018-06-14
卷宗(2018年9期)2018-06-07
分析化学(2018年12期)2018-01-22
山东工业技术(2017年14期)2017-07-18
应用化工(2014年1期)2014-08-16
