
TNF-α对肾血管收缩敏感性的影响
2013-08-24 11:53:08郭莲怡王桂君
解放军医学院学报 2013年8期
郭莲怡,王桂君,史 璇
辽宁医学院附属第一医院 消化科,辽宁锦州 121001
肝肾综合征(hepatorenal syndrome,HRS)是由于严重的肝功能损害及门脉高压所致的急性功能性肾衰竭,是终末期肝病常见的严重并发症之一,肾血管收缩是HRS的主要原因。有研究表明肝硬化时肾脏对缩血管物质的敏感性增强[1]。肿瘤坏死因子-α(tumor necrosis factor-α,TNF-α)作为重症肝炎发生的重要因子[2],与重症感染时肾衰的发生发展明显相关[3]。然而,TNF-α在HRS肾血管收缩中的作用及机制目前还不清楚。本研究应用离体灌注肾技术观察TNF-α预灌流离体大鼠肾脏后,肾血管对内皮素(endothelin,ET)收缩反应性的改变。
材料和方法
1 实验动物 选择清洁级雄性SD大鼠28只,体质量250~300 g。(由辽宁医学院实验动物中心提供,许可证号:SCXK(辽)2003-0007,开放系统饲养,实验前禁食12 h)。
2 主要试剂和仪器 TNF-α(Sigma公司)、ET-1(Sigma公司)、2-氨基乙基二苯硼酸盐(2-APB,Sigma公司)、无钙Kreb's灌流液。Minipuls2灌注泵,法国Gilson;RM6000多导生理记录仪,日本;恒温灌注池,广东;Oxnard CA张力换能器,美国Gould;氧合器,热交换器、HTG恒温器,上海;倒置显微镜,德国Carl Zeiss公司。
3 离体灌注肾实验模型的建立[4]应用戊巴比妥钠(30 mg/kg)经腹腔注射将大鼠麻醉。剪毛,腹壁消毒,正中切开,用棉签分离肠系膜上动脉、右肾动脉、右肾静脉及右侧输尿管。用套管针从肠系膜上动脉插管,经腹主动脉,插入右肾动脉起始部,管外接三通,三通接张力换能器和灌注装置,肾静脉开放。在肠系膜上动脉及右肾动脉主干分别结扎。整个手术过程均在10 min左右完成。左肾取出称重作为对照肾重。灌注过程:在腹主动脉插管至肾动脉时启动灌注泵,开始灌流。灌注液流量恒定为5 ml/min,经热交换器加温至37℃,经氧合器持续给95%氧和5%二氧化碳的混合气,灌流液为一次性,不再循环应用。
4 实验分组 28只大鼠制备离体灌注肾模型后随机分为4组(n=7),平衡期后(开始灌流5~10 min后,继续稳定20 min,共30 min),进入正式实验。具体分组:A1组(n=7):平衡期后,无钙Kreb's液灌流90 min,加ET(1 nmol/L)刺激;A2组(n=7):平衡期后,用含TNF-α(100 μg/L)的无钙Kreb's液灌流90 min,加ET(1 nmol/L)刺激;B1组(n=7):平衡期后,用无钙Kreb's液灌流90 min(其中后10 min加入30 μmol/L的2-APB),加ET(1 nmol/L)刺激;B2组(n=7):平衡期后,用含TNF-α(100 μg/L)的无钙Kreb's液灌流90 min(其中后10 min加入30 μmol/L的2-APB),加ET(1 nmol/L)刺激。
5 离体肾体外灌注过程 整个实验过程由多导生理记录仪连续记录肾脏灌注压。实验过程分三个阶段:第一阶段(平衡期):4组在灌流通路上,均先用无钙Kreb's液灌流30 min,以达到平衡;第二阶段(灌流期):各组分别用含特定成分的无钙Kreb's液灌流90 min;第三阶段(刺激期):各组均予含ET(1 nmol/L)的无钙Kreb's液继续灌流20 min。据报道离体肾体外灌流140 min内血流动力学稳定[4],故整个灌流过程持续时间为140 min。
6 肾脏水肿率及病理检查 灌流结束后,各组肾标本称重,计算肾脏水肿率,肾脏水肿率=(灌流后肾重-对照肾重)/对照肾重。4%的多聚甲醛固定24 h,石蜡包埋、HE染色观察肾小球及肾小管形态、结构。
7 统计学分析 计量资料(即各组灌注压值)以-x±s表示,各组数据比较采用两独立样本的t检验,P<0.05为有统计学意义。
结 果
1 对基础灌注压的影响 平衡期过后,各组基础灌注压比较无统计学差异(P>0.05)。TNF-α、2-APB预灌流时,基础灌注压无明显改变。
2 TNF-α对ET缩血管作用的影响 A1、A2组在应用ET(1 nmol/L)刺激后,肾灌注压较基础压均明显升高(P<0.05);A2组灌注压升高值显著高于A1组(P<0.01)。见图1,表1。
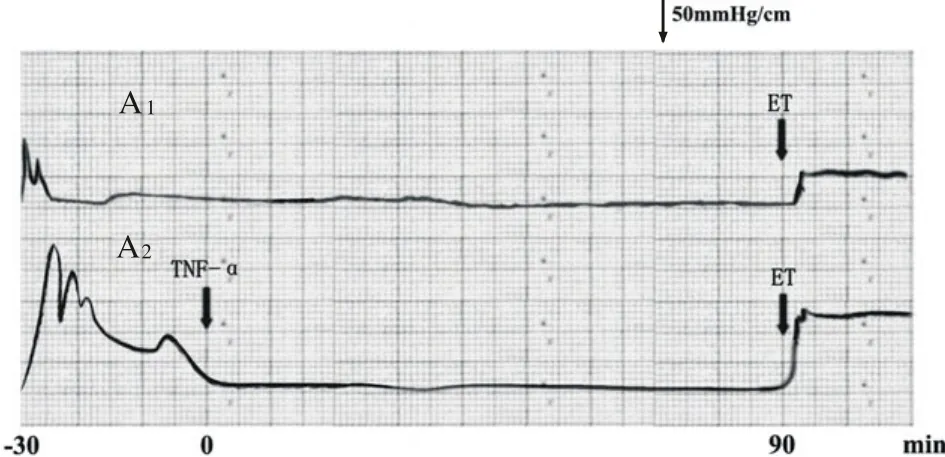
图 1 TNF-α对ET缩血管作用的影响Fig. 1 Effect of TNF-α on renal vasoconstriction by ET
3 阻断IP3R对TNF-α作用的影响 B1、B2组于灌流后期加入2-APB,刺激期用ET(1 nmol/L)刺激,肾灌注压均略升高,但与基础灌注压比较无统计学差异(P>0.05);两组灌注压升高值比较无统计学差异(P>0.05)。见图2,表1。
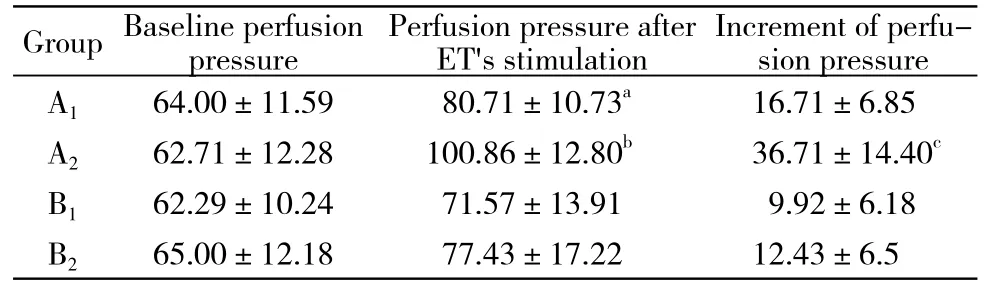
表1 各组大鼠离体肾灌注压水平Tab. 1 Renal perfusion pressure of each group(mmHg, -x±s, n=7)
4 肾脏水肿率及病理变化 4组肾脏的水肿率分别为28%±2%、29%±3%、28%±3%、27%±2%,且灌流后的肾脏标本切片经HE染色均未发现明显的器质性损伤,与灌流前相符。见图3。
讨 论
重型肝炎和各种原因引起的肝硬化常并发多脏器功能衰竭,尤其当并发HRS时,患者的预后极差,80%~100%的患者在诊断后的1个月内死亡。HRS不同于原发性肾脏疾病的肾衰竭,它缺乏后者所具有的临床、实验室和组织学的改变,目前认为HRS的发生是由于肾血管收缩造成肾脏明显低灌流的结果,其机制尚不完全清楚。当HRS发生时,患者血清中许多缩血管活性物质水平明显增高,并可直接或通过间接途径引起肾血管收缩,是导致肾血流灌注和肾功能紊乱的重要介质[1]。近年来学者们认为:重型肝炎患者血中高浓度的TNF-α是引起多脏器功能衰竭发生的重要因子[2],许多研究表明TNF-α可通过多种途径引起血管收缩,从而参与相关疾病的病理生理过程:1)TNF-α可通过TNFR2和ET-ETR依赖途径引起血管收缩。TNF-α可通过引起ET产生增多来发挥缩血管作用。当阻断ETA、、ETB受体后,TNF-α就不再有缩血管作用了[5]。2)TNF-α抑制内皮依赖的一氧化氮-环磷酸鸟苷(NO-CGMP)途径导致血管收缩。首先TNF-α通过活化蛋白激酶C(PKC)抑制内皮型一氧化氮合酶(eNOS)磷酸化从而抑制eNOS的活性;同时TNF-α可通过缩短eNOS mRNA的半衰期来下调eNOS mRNA[6]。3)TNF-α可增强Ca2+依赖及非Ca2+依赖蛋白激酶活性,增强血管平滑肌细胞收缩[7]。
本实验发现:肾脏经含TNF-α的无钙Kreb's液灌流后,其肾血管对ET的反应提高,表现为肾血管灌注压明显高于非TNF-α处理组。结果显示:TNF-α可增强ET引起的肾血管收缩。
肾血流量受血管平滑肌的收缩和舒张调节,而血管平滑肌的收缩和舒张又受平滑肌细胞内Ca2+调节。细胞内Ca2+增加受两个作用机制来调控[8]:一方面胞外Ca2+进入胞内:各种刺激,如膜电压改变、受体结合、第二信使及机械刺激都可以使细胞膜上的Ca2+通道开放,引起胞外Ca2+大量内流;另一方面胞内钙库释放Ca2+:各种刺激使肌浆网(内质网)膜上的钙释放通道开放,引起细胞内储备Ca2+的释放。本实验中应用无钙Kreb's液灌流肾脏,不存在胞外Ca2+内流这一因素,因此实验结果也表明:TNF-α可通过促进胞内钙释放进而增强ET的肾血管收缩作用。
已知ET等血管活性物质与靶细胞膜上特异性受体(如ET受体)结合后,使膜磷脂酰肌醇4,5-二磷酸发生磷酸化生成IP3,IP3与IP3R(胞内主要的Ca2+释放通道)结合则促进细胞内Ca2+释放,使胞内Ca2+水平增高[9],引起细胞收缩。2-APB是一种常用的IP3R的阻断剂,可特异性阻断IP3R[10],本实验应用含2-APB及TNF-α的无钙Kreb's液灌流肾脏,其肾血管对ET的反应较差,ET刺激后的肾血管灌注压无显著变化。结果表明:2-APB可阻断TNF-α的前述作用,说明TNF-α可能通过上调IP3R进而增强ET引起的肾血管收缩。
已有研究报道:TNF-α可诱导肾小球纤维蛋白沉积、中性粒细胞浸润、细胞凋亡,引起GFR下降。因此实验结束后,所有的肾脏标本称重,计算其水肿率<30%,说明灌流成功;并进行常规病理检查均未发现肾小管及肾小球细胞病理性改变,故排除了实验结果受器质性肾脏损伤影响的因素[11]。
综上所述,重症肝炎患者血中存在的高浓度TNF-α可能上调IP3R,促进胞内钙释放,诱导肾入球动脉平滑肌收缩性增强,肾入球动脉收缩,造成肾血流减少,参与HRS发生。
1 Leung W, Wong F. Hepatorenal syndrome: do the vasoconstrictors work?[J]. Gastroenterol Clin North Am, 2011, 40(3): 581-598.
2 Tobon GJ, Cañas C, Jaller JJ, et al. Serious liver disease induced by infliximab[J]. Clin Rheumatol, 2007, 26(4):578-581.
3 Justo Ávila P, Gracia Iguacel C, Ortiz Arduán A, et al. Acute renal failure in patient treated with anti-tumour necrosis factor alpha[J].Nefrologia, 2011, 31(4):484-488.
4 Genesca J, Segura R, Gonzalez A, et al. Nitric oxide May contribute to nocturnal hemodynamic changes in cirrhotic patients[J]. Am J Gastroenterol, 2000, 95(6): 1539-1544.
5 Donate PB, Cunha TM, Verri WA Jr, et al. Bosentan, an endothelin receptor antagonist, ameliorates collagen-induced arthritis: the role of TNF-α in the induction of endothelin system genes[J]. Inflamm Res, 2012, 61(4): 337-348.
6 Bonthius DJ, Bonthius NE, Li S, et al. The protective effect of neuronal nitric oxide synthase (nNOS) against alcohol toxicity depends upon the NO-cGMP-PKG pathway and NF-kappaB[J].Neurotoxicology, 2008, 29(6): 1080-1091.
7 Usui T, Yamawaki H, Kamibayashi M, et al. Mechanisms underlying the anti-inflammatory effects of the Ca2+/calmodulin antagonist CV-159 in cultured vascular smooth muscle cells[J]. J Pharmacol Sci,2010, 113(3): 214-223.
8 Thurley K, Skupin A, Thul R, et al. Fundamental properties of Ca2+signals[J]. Biochim Biophys Acta, 2012, 1820(8): 1185-1194.
9 Gerasimenko JV, Lur G, Ferdek P, et al. Calmodulin protects against alcohol-induced pancreatic trypsinogen activation elicited via Ca2+release through IP3 receptors[J]. Proc Natl Acad Sci U S A, 2011,108(14): 5873-5878.
10 Szatkowski C, Parys JB, Ouadid-Ahidouch H, et al. Inositol 1,4,5-trisphosphate-induced Ca2+ signalling is involved in estradiolinduced breast cancer epithelial cell growth[J]. Mol Cancer, 2010,9:156.
11 Tölle M, Schuchardt M, Wiedon A, et al. Differential effects of uridine adenosine tetraphosphate on purinoceptors in the rat isolated perfused kidney[J]. Br J Pharmacol, 2010, 161(3): 530-540.
猜你喜欢
中国生殖健康(2020年8期)2021-01-18 03:05:30
中华养生保健(2020年7期)2020-11-16 01:14:24
现代临床医学(2019年6期)2019-12-07 06:03:22
山东林业科技(2019年2期)2019-06-03 10:10:54
中国生殖健康(2018年3期)2018-11-06 07:20:10
现代园艺(2017年11期)2017-06-28 11:32:46
奥秘(2016年10期)2016-12-17 13:13:11
Coco薇(2016年8期)2016-10-09 23:51:02
实用临床医学(2016年8期)2016-06-07 01:28:18
西藏科技(2015年9期)2015-09-26 12:15:31
