
用于基因芯片数据分析的模块性图聚类方法*
2013-08-16 07:58李力曹以诚毛晓帆
华南理工大学学报(自然科学版) 2013年12期
李力 曹以诚 毛晓帆
(华南理工大学 生物科学与工程学院,广东 广州 510006)
对由节点和边组成的图进行划分得到一系列的子图,子图内的连接比较紧密而子图间的连接相对稀疏,这一划分过程称为图聚类,所得子图称为聚类[1].2008年,Kawamura 等[2]首次将图聚类方法应用在基因芯片数据分类分析中,将DNA 芯片数据使用图聚类处理后构建分类器,分类精度较二分类法提高80%,并成功在由38 个对照组样组成的分类簇中发现了胸腺癌相关基因,显示了图聚类方法在高维度数据学习和生物学对象相互关系分类方面的优势.基因相互关系网络的数学模型是一个无向图[3],图中的节点表示基因,边线表示相应基因表达谱的相似度.因此,基因表达数据的聚类问题可以简化为图论问题,这也是图聚类方法可以较好应用于基因芯片数据分析的基础.同时,基因网络还是一个特殊的复杂网络,节点数目庞大且节点之间的相互关系错综复杂[4].在这个网络中,模块是实现各种生物功能的主体,各个模块都承担着不同的生物学功能,所以越来越多的研究者使用模块识别的方法对基因芯片数据进行图聚类分析.但是,一直以来这些模块识别方法缺少一种定量的评价方法.2004年,Newman 等[5]基于协调混合(Associative Mixing)理论提出一个衡量网络模块划分质量的指标——模块性(Modularity),在这个指标下,较大的模块性值代表着对网络有一个较好的划分.此外,由于模块识别对单节点的分配过程会导致算法陷入局部最优解,Mei等[6]在模块性图聚类过程中加入了一个扰乱概率因子.加入扰乱概率因子的优点在于可以随机破坏前一次聚类过程中输出的子图结构,以搜索全局最优解;但缺点也很明显,即在多次迭代运算之后,对已输出子图结构会产生无目的的破坏,大大影响了结果的稳定性.
针对现有模块识别的定量评价方法存在的不足,文中提出了一种基于模块性指标和子图平滑度(Smoothness)的全局图聚类方法(Module Smoothness),为了防止算法陷入局部最优解,寻找最优的全局模块性,文中引入子图平滑度的定义,对每一次聚类结果产生的异常值比平均水平多的子图进行打散,再利用打散得到的单节点进行下一次聚类,多次迭代后得到具有较强图结构的基因网络的划分,即最优聚类结果.文中还利用Module Smoothness 方法对一组人外周血淋巴细胞基因组表达数据进行了分析,并将聚类结果与其他常用基因芯片数据聚类方法进行了比较.
1 材料与方法
1.1 芯片数据与预处理
从GEO 数据库[7]中选用放疗毒性与DNA 转录异常研究实验数据集GSE1725,共包含171 个人外周血淋巴细胞基因组表达数据样本,来自57 个病人,芯片平台Affymetrix Human Genome U95 V2,每个样本包含6 808 个基因表达数据[8].所有基因原始表达数据转换为对数,使用Lowess 方法[9]进行归一化处理.
1.2 图聚类
图聚类过程通常分为两步:图构造和图划分.如图1 所示,首先根据基因表达数据集计算每个表达谱之间的相关系数,即图结构的边,得到一个抽象意义上的总的基因相互关系图,图中的节点和边构成的矩阵称为模式特征矢量集;再根据合适的距离聚类算法找到基于联接的大型图中联接良好的类似小集团的最佳簇,也即具有生物学意义的相互关联的基因聚类.

图1 图聚类原理示意图Fig.1 Schematic diagram of graph clustering
1.2.1 相关系数计算
在基因表达数据的聚类分析中,通常使用皮尔森公式[10]计算基因间的相关系数.但是,该公式对正向反向共表达数据的度量是建立在统计基础上的,无法反映基因表达数据中实际具有的时序性特征.为此,文中采用了王晗[11]提出的基于打分的相似度计算方法,具体算法简要描述如下:
首先,将原始数据矩阵转换为差分矩阵,对样本总数N,整个表达谱的X 轴由样本序列s(s=1,2,…,N)变成统一的单位时间序列t(t=1,2,…,S -1),由此连接样本间不同表达量的线段与X 轴形成S-1 个夹角θt,将θt的二倍角正弦值代入式(1)中得到任意两个基因(i,j)在相邻两样本间表达变化量的相似度Gij(t).
随后,进行基因表达变化趋势的相似度计算.如式(2)所示,函数fij(t)对基因i 和基因j 在所有时间段内的变化趋势进行打分,如果两个单位时间内两基因的变化趋势一致,则该时段的打分为前一段打分的值加上常数c,如不一致则重置为初始值.
最后,将前面计算得到的表达值相似度与变化趋势相似度使用式(3)结合起来,将每个时段内基因i和基因j 的变化量差异值和对变化趋势的打分相乘,然后进行累加最后得到统一的相关系数rij(t).

式中,a、b 均为常数.

式中,rij代表基因i 和基因j 之间统一的相关系数.
1.2.2 模块性的定义
通过上述方法将基因与基因之间相关系数映射得到加权构造图G=(V,E),其中节点v(V)表示基因,e(E)表示基因表达谱间的距离.图聚类的目标是将构建的网络图划分为若干相互关系更为紧密的子图,最终使得子图内部节点相似度较高,而不同子图的节点之间相似度较低.模块性就是为了量化这种网络结构而提出的一个衡量网络模块划分的标准[5].对于一个给定了划分、由n 个节点和m 条边组成的图,其模块性(Q 值)计算公式如下:
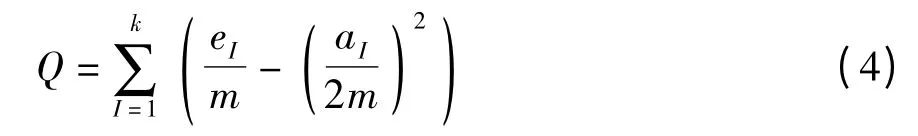
式中,k 代表子图的个数,eI是子图I 内部所有边的数目,aI是子图I 中所有节点的度之和.
1.2.3 基于平滑度的最优模块性值计算
模块性图聚类的通常做法是:先将每一个节点作为单独的子图,然后在这个初始划分Cmax的基础上,依次将每个节点移动到使Q 值增长最快的其他子图中去,直至整个网络的Q 值不能再增大为止[6].至此,所有的节点均处于较合适的子图中,但这只是相对于单节点移动的一个局部最优解.为寻求最优的全局模块性,对每次聚类得到的所有子图进行平滑度的计算.理论上关系相对集中的基因节点蔟,各节点之间的表达谱相关系数(即边线距离)应该服从正态分布的原则,其方差值越小,各节点间的关联程度就越集中,该子图的平滑度就越高,也意味着簇内基因的表达情况有更为相似的规律;反之,蔟内基因关系的异常值越多,方差值越大,子图的平滑度就越低,簇内基因的关联性较差.子图平滑度的计算方法如下.

式中,S 代表子图平滑度,m 代表所有边数,RI表示第i 个边的边长,¯R 为所有边长的算术平均值.
由此得到每个子图的平滑度S 和所有子图平滑度的平均值¯S,将平滑度低于总体平均值的子图打散成单个节点,再对所有单节点子图重复聚类过程.整个算法流程可以看作是收缩和打散步骤迭代多次求全局最优解的过程,其形式化定义如下:
Module Smoothness 算法:
1.将每一个节点作为一个单独的子图,记录当前的子图结构:Cbest和当前结构模块性值:Qbest
2.repeat /* 重复收缩和打散步骤* /
3.repeat /* 重复聚类过程* /
4.for each clustering x /* 遍历所有子图* /
6.end for
7.until Q 不再增加
8.if Q>Qbest,更新Cbest和Qbest
9.for each clustering x /* 遍历所有子图* /
10.if S <¯S,将子图x 中每一个节点作为一个单独的子图
11.end for
12.until Qbest不再增加在上述算法中,表示将节点y 合并后子图内的所有边长和增加值最大的子图α.
1.3 对比分析
为了考察Module Smoothness 方法的聚类效能,将该方法与4 种常用的基因表达数据聚类方法相比较,考察这些方法在对同一数据聚类分析过程中的类间重叠度和品质因子值.4 种常用聚类方法为:k均值聚类方法、自组织(SOM)聚类方法、Fuzzy 模糊聚类方法和经典图聚类方法(使用皮尔森公式和Ncut 算法[12]).
品质因子FOM 是Yeung 等[13]提出的用来比较不同方法聚类效能的工具,它用考察类内变异总和的方法来判断类的质量水平,从而找出最优的聚类算法.FOM 方法也能够帮助观察实际数据中的最优分类数,是评价聚类方法质量的有效工具.但是,随着聚类数的增加,FOM 值有自然增大的趋势,因此,文中使用易东等[14]改进的FOM 计算方法来考察5种聚类方法的聚类效能,改进的计算方法如下:

式中,FOM'表示改进后的品质因子,n 表示总节点数,此处代表总基因数,k 代表当前分类的簇数.
2 结果与分析
2.1 聚类性能分析
类间重叠度常用来度量不同聚类间的分离程度,对于同一组数据而言,不同聚类方法的分类结果的类间重叠度低,就说明方法的聚类表现相对优于其他方法[15].5 种算法在放疗毒性实验数据集上的类间重叠度变化如图2(a)所示,从图中可知:Module Smoothness 方法的重叠度曲线整体上比其他4 种聚类方法更靠近X 轴,聚类表现优于这些方法;随着聚类数量的增加,该方法的重叠度值变化更加稳定.
图2(b)中显示的是5 种算法在GSE1725 数据集上的FOM'变化曲线图,通过对算法的FOM'变化曲线进行比较,可以判断各聚类算法的整体效能.如图2(b)所示,Module Smoothness 方法的聚类效能总体上要优于其他4 种算法;在聚类数到达39 类之后,大多数算法的下降趋势均趋于平稳,可以认为该数据集最佳聚类数在39 左右,因此对5 种方法在聚类数为39 时的FOM'值进行比较,Module Smoothness 方法的FOM' 值比经典图聚类方法、k 均值、SOM 及Fuzzy 算法分别低28.41%、19.21%、9.84%和24.67%;此外,经典图聚类算法因为只是从统计学意义上对表达数据进行聚类,类内变异度明显大于其他方法,聚类质量在5 种方法内最差.
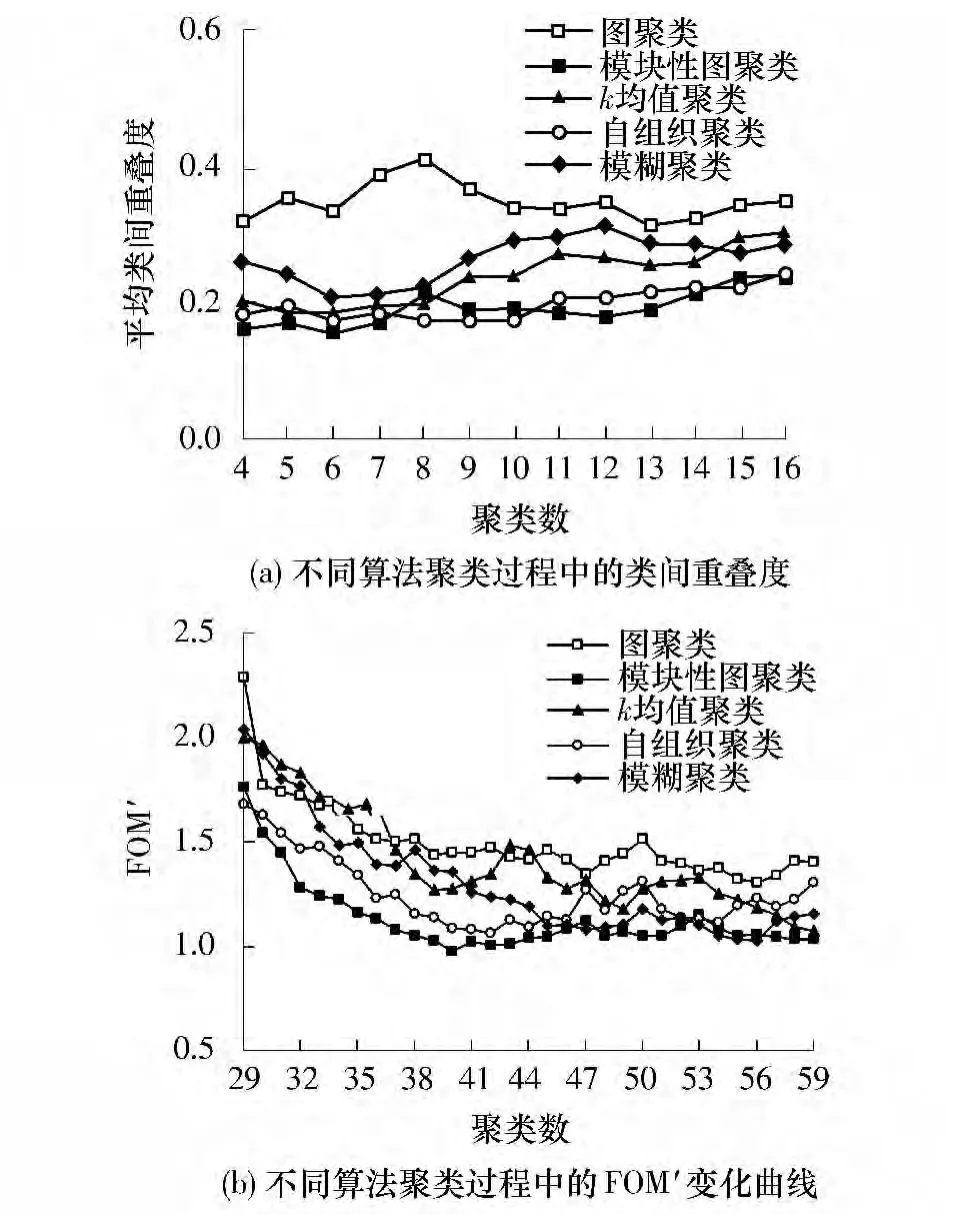
图2 不同算法聚类分析外周血淋巴细胞基因表达数据的比较Fig.2 Comparison of the Peripheral blood lymphocytes genes expression data analyzed with various clustering algorithms
2.2 聚类结果
网络智能检索系统GeneCards 是魏兹曼科学研究院建立的提供以基因为中心的信息自动整合及挖掘工具[16].为了验证Module Smoothness 算法的聚类结果,从GeneCards 网站得到与ATF-2 转录因子通路相关的77 个基因,查找这些基因的聚类结果,其中12 个基因(SMAD2,TCF3,MYC,VDR,CEBFB,CDK2,AKT1,CDK4,TGIF2,RBBP4,HDAC1,SMAD4)被正确地聚类在一起.这12 个与ATF-2 转录因子通路相关基因的聚类结果见图3,从图3 中可以看出,这些聚在同一类中的基因在样本中表达的时序变化规律非常相近.

图3 12 个与ATF-2 转录因子通路相关基因的聚类结果Fig.3 Clustering results of 12 genes related to ATF-2 transcription factor pathway
GSE1725 试验集作者Rieger 等[8]使用层次聚类的方法筛选出52 个相关基因,并按照基因功能将它们分为8 类.文中将其作为参考标准,使用性能分析结果中聚类效能较近的Module Smoothness 和自组织算法对这52 个基因进行聚类,所得3 种不同算法的聚类结果如表1 所示.

表1 3 种不同算法聚类结果1)Table 1 Clustering results of 3 different clustering algorithm
从表1 可知:自组织聚类结果与原文中的层次聚类结果相差不大,都将同属于DNA 修复功能类的RAD23B 和PSF 基因分到不同的两个类,而文中所建立的方法将RAD23B 和PSF 基因准确地聚在了同一个类中;此外,同属细胞凋亡功能的TNFSF7 和SLC25A5 基因等也得到了正确的聚类.
以上的聚类结果分析说明,Module Smoothness方法从基因芯片数据中提取到了有效的生物学信息,将具有相近生物功能和表达谱具有相似时序变化规律的基因正确地聚类到了一起,其分类准确度优于层次聚类和自组织聚类方法.
2.3 算法执行效率
基因芯片数据集中的基因数量和样本数量通常都非常庞大,分析过程中需要耗费大量的计算机资源,因而对聚类算法的执行效率提出了较高的要求.使用Module Smoothness 与前述的经典图聚类方法、k 均值、SOM 及Fuzzy 算法,在相同配置的计算机上对GSE1725 数据集的全部6808 个基因进行聚类分析,结果见表2.

表2 不同算法的聚类执行效率Table 2 Execution efficiency of various clustering algorithm
从表2 可知,除经典图聚类外,其他4 种算法聚类数都在44 左右;其中,k 均值凭借局部搜索寻找最优解方法完成聚类,所耗时间最少,而Module Smoothness 在保证聚类结果的条件下,执行效率和SOM 算法相近,比Fuzzy 算法高5.94%.
3 结语
文中提出了一种基于模块性指标和子图平滑度的全局图聚类方法Module Smoothness,为了防止算法陷入局部最优解,寻找最优的全局模块性,引入子图平滑度的定义,对每一次聚类结果产生的异常值比平均水平多的子图进行打散,再利用打散得到的单节点进行下一次聚类,多次迭代后得到具有较强图结构的基因网络的划分,即最优的聚类结果.利用该方法与经典图聚类、k 均值、SOM 及Fuzzy 4 种常用的基因芯片数据聚类方法,对一组人外周血淋巴细胞基因组表达数据进行了分析比较.结果表明:该方法在聚类过程中的平均类间重叠度和FOM'值总体上优于其他4 种算法,在将数据集分类到最佳聚类数39 时,其FOM' 值分别比上述4 种方法低28.41%、19.21%、9.84% 和24.67%;聚类结果的生物信息学分析显示,该方法将具有相近生物功能和表达谱具有相似时序变化规律的基因准确地聚类到了一起,其分类准确度高于层次聚类和自组织聚类方法;在算法的执行效率方面,Module Smoothness在保证聚类结果的条件下,执行效率和SOM 算法相近,比Fuzzy 算法高5.94%.
[1]邓健爽,郑启伦,彭宏,等.基于连通图动态分裂的聚类算法[J].华南理工大学学报:自然科学版,2007,35(1):118-122.Deng Jian-shuang,Zeng Qi-lun,Peng Hong,et al.Clustering algorithm based on dynamic division of connected graph[J].Journal of South China University of Technology:Natural Science Edition,2007,35(1):118-122.
[2]Kawamura T,Mutoh H,Tomita Y,et al.Cancer DNA microarray analysis considering multi-subclass with graphbased clustering method [J].Journal of Bioscience and Bioengineering,2008,106(5):442-448.
[3]Rapaport F,Zinovyev A,Dutreix M,et al.Classification of microarray data using gene networks[J].BMC Bioinformatics,2007,8(1):35.
[4]Guimerà R,Sales-Pardo M,Amaral L A N.Module identification in bipartite and directed networks[J].Physical Review E,2007,76(3):036102.
[5]Newman M E J,Girvan M.Finding and evaluating community structure in networks [J].Physical Review E,2004,69(2):026113.
[6]Mei J,He S,Shi G,et al.Revealing network communities through modularity maximization by a contraction-dilation method[J].New Journal of Physics,2009,11(4):1-14.
[7]Barrett T,Troup D B,Wilhite S E,et al.NCBI GEO:mining tens of millions of expression profiles:database and tools update[J].Nucleic Acids Research,2007,35:760-765.
[8]Rieger K E,Hong W J,Tusher V G,et al.Toxicity from radiation therapy associated with abnormal transcriptional responses to DNA damage[J].PNAS,2004,101(17):6635-6640.
[9]Yang Y H,Dudoit S,Luu P,et al.Normalization for cDNA microarray data:a robust composite method addressing single and multiple slide systematic variation[J].Nucleic Acids Research,2002,30(4):15-e15.
[10]Guzzi P H,Veltri P,Cannataro M.Unraveling multiple miRNA-mRNA associations through a graph-based approach[J].ACM-BCB,2012(10):649-654.
[11]王晗.整合变化量与变化趋势的共调控基因相似性度量[D].长春:吉林大学计算机科学与技术学院,2008.
[12]Shi X,Su S,Long J,et al.MicroRNA-191 targets Ndeacetylase/N-sulfotransferase 1 and promotes cell growth in human gastric carcinoma cell line MGC803[J].Acta Biochim Biophys Sin (Shanghai),2011,43(11):849-856.
[13]Yeung K Y,Haynor D R,Ruzzo W L.Validating clustering for gene expression data[J].Bioinformatics,2001,17(4):309-318.
[14]易东,杨梦苏,李辉智.基因表达数据聚类分析结果的评价方法研究[J].中国卫生统计,2002,19(6):332-335.Yi Dong,Yang Meng-su,Li Hui-zhi.An nuvel method for evaluation of clustering results for gene expression data[J].Chinese Journal of Health Statistics,2002,19(6):332-335.
[15]Datta S,Datta S.Comparisons and validation of statistical clustering techniques for microarray gene expression data[J].Bioinformatics,2003,19(4):459-466.
[16]Belinky F,Bahir I,Stelzer G,et al.Non-redundant compendium of human ncRNA genes in GeneCards [J].Bioinformatics,2013,29(2):255-261.
猜你喜欢
机械工业标准化与质量(2022年6期)2022-08-12
国际眼科杂志(2021年9期)2021-09-15
装备制造技术(2020年2期)2020-12-14
铁道通信信号(2019年6期)2019-10-08
同济大学学报(自然科学版)(2019年2期)2019-04-02
雷达学报(2017年6期)2017-03-26
电子科技大学学报(2016年2期)2016-08-31
中国卫生(2015年12期)2015-11-10
电子设计工程(2015年6期)2015-02-27
华东师范大学学报(自然科学版)(2014年1期)2014-04-16
