
高灵敏度测定壳聚糖酶活力的新方法及其比较研究
2013-08-07 09:02张永勤常海燕刘征东罗彩华牟晓凤
食品科学 2013年9期
张永勤,张 杰,常海燕,张 坤,刘征东,罗彩华,牟晓凤
(青岛科技大学化工学院,山东 青岛 266042)
近年来,随着低聚壳聚糖以及壳寡糖在医药、生物材料、生物能源、食品工业、化妆品以及环境保护等诸多领域的不断应用,壳聚糖水解酶已成为国内外诸多学者的研究热点[1-5]。在壳聚糖的水解工艺中,与传统化学水解法相比,酶解法更易控制,底物特异性强,副产物少。但是,由于壳聚糖酶的活力测定方法普遍具有较低的灵敏度,一方面影响了壳聚糖酶产生菌的筛选进程,致使壳聚糖酶的价格居高不下[6],另一方面也限制了壳聚糖酶在应用过程中的质量控制。酶活力是衡量水解酶性能的关键指标,酶活力测定方法的研究,能够为壳聚糖水解酶在筛选、生产和应用过程中提供有效的质量控制手段。因此,寻求一种有效的壳聚糖水解酶活力测定方法对于壳聚糖产业发展十分重要。目前常用Somogyi-Nelson法[7]、3,5-二硝基水杨酸法(DNS法)[8]、铁氰化钾法[9]和BCA法[10-11]测定多糖水解酶活力,但由于这些方法或多或少存在灵敏度、测定范围、试剂高毒性、蛋白质影响等问题导致了其在多糖水解酶活力测定的应用上受到不同程度的限制[12]。
本实验将3-甲基-2-苯并噻唑啉酮腙盐酸盐水合物(MBTH)高灵敏度测定还原糖含量的方法[13-14]应用到壳聚糖酶的活力测定中,以高脱乙酰壳聚糖为酶解底物,并针对壳聚糖本身的物化特性设计并优化活力测定方法。近年来有学者报道了果胶酶[15]、木瓜蛋白酶[16]、蛋白酶[17]、溶菌酶[18]以及纤维素酶[6,19-20]等都具有壳聚糖水解酶活力,特别是纤维素酶具有的壳聚糖水解酶活力较高[21]。考虑到壳聚糖酶的纯酶制剂价格昂贵,因此,本研究以含有壳聚糖酶活力的纤维素酶制剂为实验材料,利用MBTH法测定其所含有的微量壳聚糖水解酶活力,并与目前最常采用的DNS法和铁氰化钾法做对比,以期对壳聚糖酶的定量、筛选与应用提供帮助。
1 材料与方法
1.1 材料、试剂与仪器
纤维素酶(ROCKSOFTTM ACE P150) 日本Dyadic公司;壳聚糖(93.5%脱乙酰度,1H NMR) 实验室自制;氨基葡萄糖、3-甲基-2-苯并噻唑啉酮腙盐酸盐水合物(MBTH)、二硫苏糖醇(DTT) 美国Sigma公司;其他试剂均为分析纯。
SHZ-82恒温水浴振荡器 常州智博瑞仪器制造有限公司;UNICO-2100紫外-可见分光光度计 尤尼科(上海)仪器有限公司;pH计 梅特勒-托利多(上海)仪器有限公司;移液器 赛默飞世尔(上海)仪器有限公司。
1.2 方法
1.2.1 壳聚糖溶液的配制
称取2.000g壳聚糖于500mL烧杯内,逐滴加入10mL 0.2mol/L醋酸溶液,搅拌至壳聚糖完全溶解,且体系呈透明凝胶状,在4℃冰箱静置2h。加入适量0.2mol/L醋酸钠溶液,搅拌至胶状体系溶解,调节pH值至6.0,适当时可加微量10mol/L氢氧化钠溶液调节溶液pH值。将溶液转移至500mL容量瓶内,用0.2mol/L、pH6.0醋酸-醋酸钠缓冲溶液定容至500mL,充分混匀。配制成4mg/mL的壳聚糖溶液于4℃冰箱内贮藏备用。整个酶解体系使用0.2mol/mL、pH6.0醋酸-醋酸钠缓冲溶液为溶剂,以下同。
1.2.2 氨基葡萄糖标准曲线的绘制
MBTH法:用醋酸-醋酸钠缓冲溶液分别配制终质量浓度为0~80μg/mL的氨基葡萄糖溶液(每个稀释度各含有2mg/mL壳聚糖底物),取2mL该溶液与2mL 0.5mol/L氢氧化钠溶液充分混匀,8000r/min离心5min,取1mL上清液与0.5mL MBTH试剂充分混匀,于80℃水浴15min,迅速加入1mL硫酸铁铵试剂(0.5g/100mL (FeNH4(SO4)2)·12H2O、0.5%氨基磺酸、0.5mol/L盐酸),混匀,于655nm波长处测定吸光度,每组3个平行样。
DNS法:参照Miller[8]的方法,并根据本实验壳聚糖酶活力测定方法的特点稍加修改。用醋酸-醋酸钠缓冲溶液配制终质量浓度为0~1.5mg/mL的氨基葡萄糖溶液(每个稀释度各含有2mg/mL壳聚糖底物)。取2mL还原糖溶液与0.2mL 10mol/L氢氧化钠溶液充分混匀,8000r/min离心5min,取0.4mL上清液向其中加入0.3mL DNS试剂,沸水浴15min,迅速冷却,加入4.3mL蒸馏水中,充分混匀,于500nm波长处测定吸光度值,每组3个平行样。
铁氰化钾法:参照文献[9]的方法,并根据本实验壳聚糖酶活力测定方法的特点稍加修改。用醋酸-醋酸钠缓冲溶液配制终质量浓度为0~100μg/mL的氨基葡萄糖溶液(每个稀释度各含有2mg/mL的壳聚糖底物)。取4mL还原糖溶液加入0.4mL 10mol/L氢氧化钠溶液,充分混匀,8000r/min离心5min,取1.2mL上清液与1.6mL铁氰化钾试剂充分混匀,沸水浴15min,迅速冷却,于420nm波长处测定吸光度,每组3个平行样。
1.2.3 酶活力测定方法
多点法(罐法、动力学法、动态法):在锥形瓶中将已预热的底物溶液与酶溶液迅速充分混匀(MBTH:19.8mL底物+0.2mL酶溶液,DNS:10mL底物+10mL酶溶液,铁氰化钾:36.4mL底物+0.4mL酶溶液;底物终质量浓度均为2mg/mL),在37℃条件下水浴加热,分别在0、10、20、30、40、50、60min时取酶解液与氢氧化钠充分混合终止反应(MBTH:2mL酶解液+2mL 0.5mol/L氢氧化钠,DNS:2mL酶解液+0.2mL 10mol/L氢氧化钠,铁氰化钾法:4mL酶解液+0.4mL 10mol/L氢氧化钠),8000r/min离心15min,取上清液,按照1.2.2节中的方法测定酶解液中还原糖含量,每组做3个平行样。
终点法(试管法):在试管中将已预热的1mL 4mg/mL底物溶液与1mL酶溶液充分混匀(底物终质量浓度为2mg/mL),在37℃条件下水浴加热反应1h,加入氢氧化钠终止反应(各方法中氢氧化钠浓度与比例同上),8000r/min离心15min,取上清,按照1.2.2节中的方法测定酶解液中还原糖含量,每组做3个平行样。
酶活力单位(1U)定义:在上述MBTH法的反应条件下,使每毫升酶解液每分钟产生相当于1μmol氨基葡萄糖的还原糖的量所需的酶量为一个酶活力单位。检测限(DL)和定量限(QL)根据公式(1)和(2)[22]计算所得。

式中:Sbi是9个空白溶液的标准偏差;b为回归方程的斜率。
1.2.4 酶动力学曲线的测定方法
将纤维素酶溶液与壳聚糖底物溶液按一定比例混合,在37℃条件下水浴加热,分别在0、10、20、30、40、50、60min时终止反应,酶解液在8000r/min条件下离心15min,按照1.2.2节的方法测定还原糖含量,每个样品组做3个平行样。
1.2.5 底物质量浓度对酶活力测定的影响
分别配制质量浓度为0.05、0.10、0.15、0.20、0.25、0.30、0.40、0.50、0.80mg/mL的壳聚糖底物溶液,取0.2mL酶溶液与19.8mL底物溶液在试管(100mm×15mm)中混合,在37℃条件下水浴加热,分别于0、10、20、30、40、50、60min时终止反应。取酶解液在8000r/min条件下离心15min,按照1.2.2节的方法测定还原糖含量,每个样品组做3个平行样。
1.2.6 酶质量浓度曲线的绘制
按照1.2.3节的多点测定法进行酶活力测定,其中纤维素酶溶液用0.2mol/L、pH6.0醋酸-醋酸钠溶液配制, 并相应稀释8个质量浓度梯度,使酶解液中纤维素酶浓度分别为0~150μg/mL(MBTH法)、0~1.2mg/mL(DNS法)、 0~100μg/mL(铁氰化钾法)。检测限和定量限参照1.2.3节计算。
2 结果与分析
2.1 MBTH法测定氨基葡萄糖的标准曲线及其比较

图 1 3种方法测定氨基葡萄糖标准曲线(MBTH、铁氰化钾、DNS)Fig.1 Standard curves of glucosamine for the three method, MBTH, Schales’ Procedure and DNS methods
由图1可知,在测定氨基葡萄糖浓度时,MBTH法灵敏度最高,其检测限为4.5μmol/mL,而DNS法和铁氰化钾法分别为320μmol/mL和8.0μmol/mL。在DNS法中,氨基葡萄糖浓度较低时,无法检测出溶液中的还原糖含量,从而导致其标准曲线很难过原点[7-8],而在3.8mmol/mL以上的浓度点回归而成的标准曲线更接近原点,如图1中的嵌入图所示。铁氰化钾法属减色法,空白的吸光度决定着该方法的检测范围,其空白值的测定误差也决定着还原糖的检测限。当还原糖浓度超过340μmol/mL时,随着还原糖浓度的增高吸光度呈非线性变化,超出了该法的检测范围,即样品吸光度趋向零,无法测出还原糖含量,从而导致该法工作浓度范围过于狭窄。相比之下,MBTH法避开了上述缺点,其灵敏度是铁氰化钾法的1.8倍,DNS法的71倍,具有较宽的检测范围,特别适合于微量还原糖浓度的测定。
2.2 酶活力测定方法的建立及底物质量浓度对酶活力测定的影响
考虑到本实验所使用的纤维素酶在pH6.0(最适pH值)和55℃(最适温度)条件下预热15min,其酶活力即损失50%[23];壳聚糖在较高温度条件下会发生非酶降解;过低的酶解温度难以在一般实验室温度条件下得以控制,因此,本实验将酶解温度设定在37℃。由于壳聚糖不溶于碱性环境,当加入NaOH终止反应时会产生大量沉淀,需在离心后取上清液进行还原端基浓度的测定。在理论上,酶解反应中底物质量浓度越高,酶解反应速率越能维持在最大值。但是,由于过高的底物浓度具有较高黏度而影响酶解进程(传质速率较慢);在酶解终止后会产生大量沉淀而减少上清液体积;会使底物本身的还原基团浓度增加而导致本底值(即空白值)增加;且会增加测定成本等,因此,选择合适的底物浓度有助于该酶活力的准确测定。
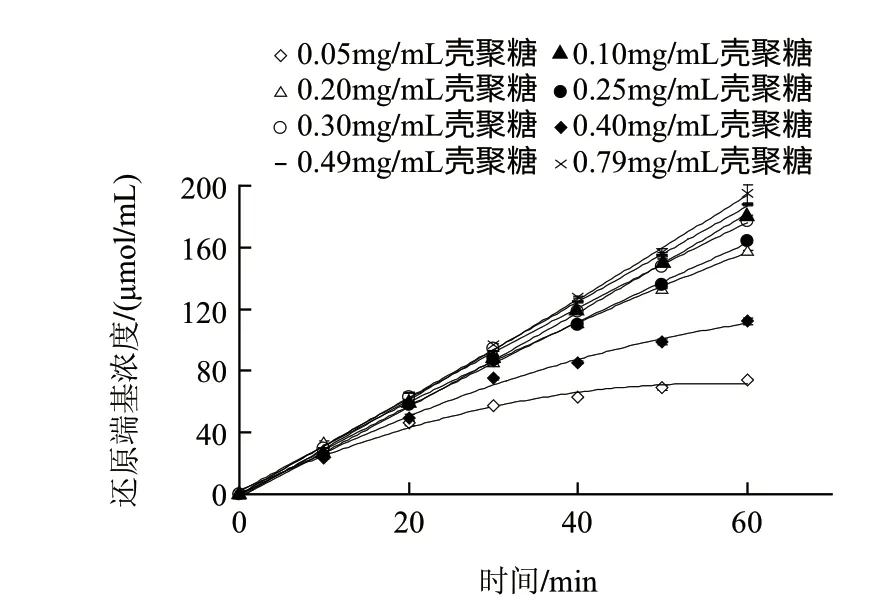
图 2 不同底物质量浓度条件下MBTH法动力学曲线Fig.2 Kinetic curves of various substrate concentrations for MBTH method
图2为在不同壳聚糖底物质量浓度条件下的酶解进程曲线。在该图中,当底物质量浓度在0.05、0.10mg/mL时,酶解反应速率分别在20、30min内保持恒定,随后酶解反应速率逐渐降低,即产物生成量与酶解时间呈非线性关系,这可能是因为随着酶解反应的进行,底物壳聚糖被不断消耗而释放出还原端基,使酶不再与底物达到饱和,或者是反应体系中不断增加的还原端基造成的产物抑制作用而导致了反应速率的下降。
当底物质量浓度高于0.20mg/mL及以上时,酶解反应速率与酶解时间在整个酶解过程中始终保持线性增加关系。另外,在0.05mg/mL时,酶解反应20min内的产物生成量只有46μmol/mL,而动力学参数如Km值的测定往往需要测定底物质量浓度在Km值附近的酶解反应速率。因此,MBTH法的高灵敏度足够用来测定动力学参数。然而,由于DNS法的灵敏度较低,其氨基葡萄糖的检测限为320μmol/mL,很难检测到产物还原端基浓度在200μmol/mL以下的酶解速率变化(图2),而增加酶浓度确实可以提高酶解速率从而增加产物浓度,但又会使酶解反应背离零级反应而很难测得酶解初速率,此外,在高底物浓度条件下,酶解速率变化不明显(图3),会引入较大测量误差,因此,用DNS法较难准确测定Km值。在图3的整个酶解过程中,随着底物质量浓度的增加,酶解反应速率逐渐递增。在低底物质量浓度范围内,反应速率与底物浓度呈线性增加关系。在底物质量浓度增加到0.30mg/mL之后,反应速率增加缓慢并最终趋于平缓。根据图3双倒数曲线公式为y=0.0303x+0.2494(R2=0.9957)计算可得Km为0.12mg/mL,遵循底物质量浓度可尽量大,但又不影响测定的原则,因此,本实验选择2mg/mL(16.7倍的Km值)作为酶活力测定的底物终质量浓度。
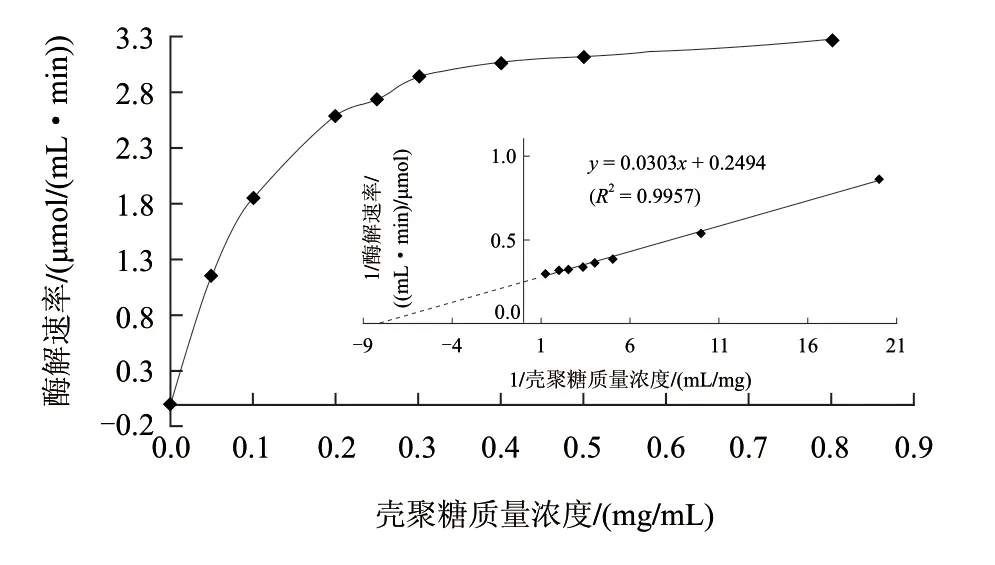
图 3 底物质量浓度与反应速率的关系和双倒数曲线图(MBTH法)Fig.3 Relationship between substrate concentration and reaction rate and Double reciprocal Lineweaver-Burk plots in MBTH method
2.3 MBTH法动态测定壳聚糖酶活力及其比较
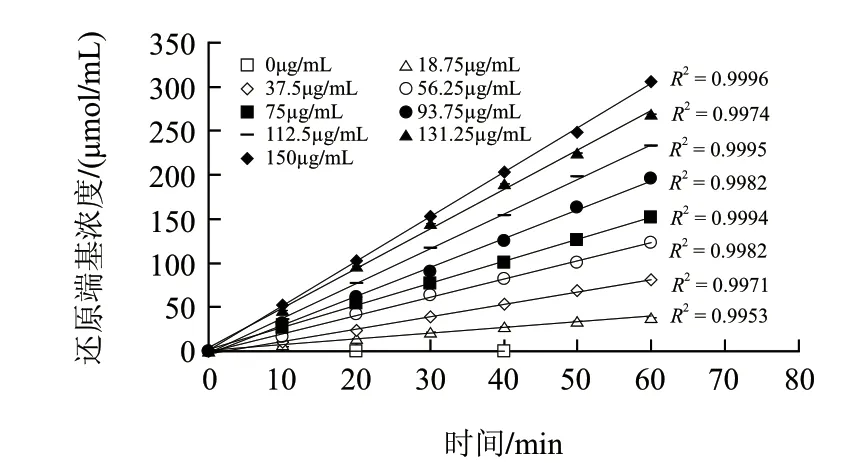
图 4 不同酶质量浓度条件下MBTH法动力学曲线Fig.4 Kinetic curves of various enzyme concentrations for MBTH method
由图4可知,不同的酶质量浓度条件下,酶解反应速率(即单位时间内产物生成量)随时间呈线性增加关系。这一线性关系说明,在不同的酶质量浓度条件下,酶解反应均达到最大反应速率,并且在60min内保持恒定。另外,只有当底物用量足以在60min内使底物与酶的结合始终保持饱和状态,才能保证酶解反应速率恒定在最大值。因此,可以说明实验中所使用的底物质量浓度要远大于酶对底物的Km值。在这样的底物质量浓度条件下,反应速率会随着酶质量浓度增加成比例增加。在测定酶活力时,只有保证在测定过程中,酶解反应速率始终保持在一个恒定的最大值,才能确保酶活力测定的准确性。因此,在测定酶活力时,选择在酶解反应60min以内测定酶活力是比较准确的,因此,按照1.2.3节中的试管法即可以代替操作繁琐的动力学法用于常规酶活力测定。

图 5 不同酶质量浓度条件下铁氰化钾法动力学曲线Fig.5 Kinetic curves of various enzyme concentrations for potassium ferricyanide method

图 6 MBTH法、铁氰化钾法和DNS法测定酶质量浓度曲线Fig.6 Enzyme concentration curves for MBTH, DNS and potassium ferricyanide method
铁氰化钾法测壳聚糖酶活力的动力学曲线如图5所示,其结果接近MBTH法,除在2.1节中所讨论的由于该法属减色法使检测范围较窄之外,较大的空白值也会使该酶活力的检测限偏高,而且,由于该法取样量较大,不利于在线监测。另外,由于壳聚糖本身对铁氰化钾有一定吸附作用,因此,终止反应后需马上离心取上清液。尽管铁氰化钾法有诸多缺陷,但是,由图6可知,其酶活力测定结果与MBTH法相近,以MBTH法的酶活力定义为基准,其检测限为20mU/mL,是MBTH法(13mU/mL)的1.6倍。
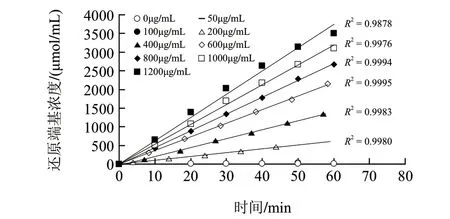
图 7 不同酶质量浓度条件下DNS法动力学曲线Fig.7 Kinetic curves of various enzyme concentrations for DNS method
而对于DNS法而言,以MBTH法的酶活力定义为基准,其检测限为1.5U/mL,是MBTH法的116倍。如图6和图7所示,当酶活力单位在1.5~21.6U/mL以内时,酶解反应速率与酶质量浓度呈线性增加关系;当酶解速率超过21.6U/mL时,尽管在不同酶质量浓度条件下的酶动力学曲线仍各呈线性递增,但是,随着酶质量浓度的增加,酶解反应速率逐渐处于非线性递增,最终曲线趋于平缓。这主要是因为酶浓度过高,底物消耗过多,酶解产物导致产物抑制。另外,之所以动力学曲线仍呈线性递增,是由于DNS法所测还原端基的摩尔吸光系数与酶解产物的聚合度密切相关,而且,聚合度越低摩尔吸光系数越大[24-26],从而造成线性动力学曲线的假象,致使酶质量浓度线性部分斜率(y=0.0641x-2.6419)超出铁氰化钾法法(y=0.0337x+0.0631)和MBTH法(y=0.0321x+0.0407)1倍之多,因此,DNS法测定壳聚糖酶活力是不准确的,它不适于生产中的质量控制、销售等环节中的准确定量。
3 结 论
建立了一种新的高灵敏度测定壳聚糖酶活力的方法——MBTH法,利用动力学法研究了该活力测定方法的测定参数,在37℃、pH6.0反应条件下壳聚糖底物溶液的终质量浓度需在2mg/mL以上,酶解时间在60min以内。MBTH法准确、灵敏、检测范围宽、操作简便,可以用终点法定量测得壳聚糖酶活力,且可用于DNS法较难测得的较小Km值的测定。该法测定结果近似于铁氰化钾法,DNS法广泛用于壳聚糖酶的筛选、分离纯化、应用中的工艺优化,但不适于质量控制、销售等环节中的准确定量。
[1] TOKORO A, TATEWAKI N, SUZUKI K, et al. Growth-Inhibitory effect of hexa-N-acetylchitohexanse and chitohexaose against meth: a solid tumor[J]. Chemical & Pharmaceutical Bulletin, 1988, 36(2): 784-790.
[2] SHAHIDI F, ARACHCHI J K V, JEON Y J. Food applications of chitin and chitosans[J]. Trends in Food Science & Technology, 1999, 10(2): 37-51.
[3] INOKUMA K, TAKANO M, HOSHINO K. Direct ethanol production from N-acetylglucosamine and chitin substrates by mucor species[J]. Biochemical Engineering Journal, 2013, 72: 24-32.
[4] KUMAR M N V R. A review of chitin and chitosan applications[J]. Reactive and Functional Polymers, 2000, 46(1): 1-27.
[5] TSAI Guojane, WU Zenyuon, SU Wenhuey. Antibacterial activity of a chitooligosaccharide mixture prepared by cellulase digestion of shrimp chitosan and its application to milk preservation[J]. Journal of Food Protection, 2000, 63(6): 747-752.
[6] LIU Jing, XIA Wenshui. Purification and characterization of a bifunctional enzyme with chitosanase and cellulase activity from commercial cellulose[J]. Biochemical Engineering Journal, 2006, 30(1): 82-87.
[7] SOMOGYI M. A new reagent for the determination of sugars[J]. Journal of Biological Chemistry, 1945, 160: 61-68.
[8] MILLER G. Use of dinitrosalicylic acid reagent for determination of reducing sugar[J]. Analytical Chemistry, 1959, 31(3): 426-436.
[9] IMOTO T, YAGISHITA K. A simple activity measurement of lysozyme[J]. Agricultural and Biological Chemistry, 1971, 35(7): 1154-1156.
[10] WAFFENSCHMIDT S, JAENICKE L. Assay of reducing sugars in the nanomole range with 2,2’-bicinchoninate[J]. Analytical Biochemistry, 1987, 165(2): 337-340.
[11] GARCIA E, JOHNSTON D, WHITAKER J R, et al. Assessment of endo-1,4-β-D-glucanase activity by a rapid colorimetric assay using disodium 2,2’-bicinchoninate[J]. Journal of Food Biochemistry, 1993, 17(3): 135-145.
[12] 张永勤, 薛长湖, 汤浩源, 等. 还原糖的可见分光光度法研究进展[J]. 食品与发酵工业, 2007, 33(5): 97-104.
[13] ANTHON G E, BARRETT D M. Determination of reducing sugars with 3-methyl-2-benzothiazolinonehydrazone[J]. Analytical Biochemistry, 2002, 305(2): 287-289.
[14] HORN S J, EIJSINK V G. A reliable reducing end assay for chitooligosaccharides[J]. Carbohydrate Polymers, 2004, 56(1): 35-39.
[15] KITTUR F S, KUMAR A B V, THARANATHAN R N. Low molecular weight chitosans: preparation by depolymerization with Aspergillus niger pectinase, and characterization[J]. Carbohydrate Research, 2003, 338(12): 1283-1290.
[16] RONCAL T, OVIEDO A, ARMENTIA I L D, et al. High yield production of monomer-free chitosan oligosaccharides by pepsin catalyzed hydrolysis of a high deacetylation degree chitosan[J]. Carbohydrate Research, 2007, 342(18): 2750-2756.
[17] LI Jin, DU Yumin, LIANG Hongbo. Infl uence of molecular parameters on the degradation of chitosan by a commercial enzyme[J]. Polymer Degradation and Stability, 2007, 92(3): 515-524.
[18] ZHANG Yongqin, WANG Zheping, ZHANG Jie, et al. Quantitative determination of chitinolytic activity of lysozyme using halfdeacetylated chitosan as a substrate[J]. Carbohydrate Polymers, 2011, 85(3): 554-559.
[19] PANTALEONE D, YALPANI M, SCOLLAR M. Unusual susceptibility of chitosan to enzymic hydrolysis[J]. Carbohydrate Research, 1992, 237(1): 325-332.
[20] ZHANG Yongqin, ZHANG Xiaoyang, DING Ruoran, et al. Determination of the degree of deacetylation of chitosan by potentiometric titration preceded by enzymatic pretreatment[J]. Carbohydrate Polymers, 2011, 83(2): 813-817.
[21] XIA Wenshui, LIU Ping, LIU Jing. Advance in chitosan hydrolysis by non-specifi c cellulases[J]. Bioresource Technology, 2008, 99(15): 6751-6762.
[22] MILLER J N. Basic statistical methods for analytical chemistry Part 2. Calibration and regression rethods a review[J]. Analyst, 1991, 116: 3-14.
[23] 张永勤. 强制渗透法制备水溶性壳聚糖及其固定化酶解方法与产物研究[D]. 青岛: 中国海洋大学, 2005.
[24] BREUIL C, SADDLER J N. Comparison of the 3,5-dinitrosalicylic acid and nelson-somogyi methods of assaying for reducing sugars and determining cellulase activity[J]. Enzyme and Microbial Technology, 1985, 7: 327-332.
[25] SENGUPTA S, JANA M L, SENGUPTA D. A note on the estimation of microbial glycosidase activities by dinitrosalicylic acid reagent[J]. Applied Microbiol Biotechnology, 2000, 53: 732-735.
[26] SAQIB A A N, WHITNEY P J. Differential behaviour of the dinitrosalicylic acid (DNS) reagent towards mono- and di-saccharide sugars[J]. Biomass and Bioenergy, 2011, 35: 4748-4750.
猜你喜欢
理化检验-化学分册(2023年1期)2023-03-20
云南化工(2021年6期)2021-12-21
山西农业大学学报(自然科学版)(2021年2期)2021-04-26
人民黄河(2020年12期)2020-12-30
山西农业科学(2020年9期)2020-09-14
科学(2020年2期)2020-08-24
山西农业科学(2019年12期)2019-12-19
电子制作(2017年10期)2017-04-18
化工技术与开发(2016年2期)2016-07-30
山西建筑(2014年6期)2014-11-09
