
高效液相色谱 串联质谱法同时测定茶叶中290种农药残留组分
2013-07-13 11:12黄峻榕郑月明储晓刚
分析测试学报 2013年1期
贾 玮,黄峻榕,凌 云,冯 峰,郑月明,储晓刚
(1.陕西科技大学 生命科学与工程学院,陕西 西安 710021;2.中国检验检疫科学研究院,北京 100123)
茶叶是不可缺少的健康饮品,也是我国的重要出口农产品,欧洲、日本是我国茶叶的重要进口国。茶叶进出口国在制定茶叶标准时需综合考虑茶叶食品安全和商品进出口两个方面。欧盟和日本作为农产品进口国,制定的许多农药最大残留标准都非常严格。国际食品法典委员会(CAC)、欧盟、日本规定茶叶中最高农药残留限量(MRL)分别为0.10~50.0、0.01~30.0、0.01~30.0 mg/kg,没有明确规定残留限量的均默认为0.01 mg/kg[1]。
茶叶中农药残留的常用检测方法包括酶联免疫分析法(ELISA)[2]、气相色谱法(GC)[3-4]、气相色谱 -质谱法(GC -MS)[5-7]、液相色谱法(HPLC)[8]、液相色谱 - 质谱法(LC - MS)[9-10]、液相色谱 -串联质谱法(LC-MS/MS)[11-14]等。ELISA、GC和HPLC的方法灵敏度较低,选择性和特异性较差,不适于多种农残痕量分析的要求。GC-MS和LC-MS虽然方法灵敏度和特异性较高,但由于难以完全阐明化合物的结构裂解信息,定性准确度方面尚有欠缺,容易产生假阳性结果。而液相色谱-串联质谱法(LC-MS/MS)因具有灵敏、准确和快速等特点,适用于分析基质复杂、背景干扰严重的痕量化合物,同时可在碰撞诱导解离模式下,得到其碎片离子的分子质量,从而进一步对化合物的结构和裂解规律加以确证,定性准确性高。目前,尚未有应用LC-MS/MS法单针进样同时分析茶叶中200多种农药的文献报道。
茶叶与其他植物源性食品相比,富含茶多酚类和色素类化合物,经不同工艺加工的茶叶产品成分也有很大差异。在农药残留测定时,不同的成分造成的干扰也会有所不同。因而其农残检测净化方式多采用传统方法,但这些前处理技术过程复杂,步骤繁琐,试剂用量大,检测成本高,干扰较大且不能满足大批量样品多组分快速测定要求。且针对四类茶叶样品,同时采用3个版本的QuEChERS前处理技术(2003 Oringinal,AOAC 2007.12008 CEN 15662)进行比较并优化尚未见报道[15-17]。
本研究采用高效液相色谱-串联质谱联用技术,针对四类茶叶样品,同时对3个版本的QuECh-ERS前处理技术进行比较,并优化了色谱质谱条件,以改进的QuEChERS方法为前处理手段,建立了茶叶中290种农药残留的定性确证和定量测定方法。
1 实验部分
1.1 仪器与试剂
TSQ Quantum Ultra高效液相色谱-串联四极杆质谱仪(美国Thermo Fisher Scientific公司);高速组织捣碎机(美的);Avnti J-26×PI型高速冷冻离心机(美国Beckman Coulter公司);Vortex.Genie2T旋涡混合器(美国Scientific Industries);振荡器(日本Yamata SA31);超声波清洗器(江苏昆山超声仪器有限公司);微孔滤膜(0.20 μm,美国Pall公司);真空氮气吹干仪(美国Caliper公司)。
甲醇、乙腈(HPLC级,美国Thermo Fisher Scientific公司);C18吸附剂、N-丙基乙二胺吸附剂(PSA,粒径范围40~60 μm,平均孔径100 ,美国安捷伦科技有限公司);石墨化炭黑(GCB,粒径范围40~120 μm,美国Supelco公司);无水硫酸钠、无水硫酸镁、氯化钠、无水醋酸钠(优级纯,上海国药集团),500℃灼烧4 h,置于干燥器中备用;倍半水合柠檬酸二钠、二水合柠檬酸钠(优级纯,美国Sigma公司);乙酸、甲酸、甲酸铵、甲苯(色谱纯,美国Sigma公司)。实验用水均为高纯水(经Milli-Q超纯水器纯化)。
290种农药标准品均购于Dr.Ehrenstorfer公司。
标准溶液的配制:分别准确称适量的农药标准品,用甲醇制成1 000.0 mg/L单一标准储备液,于-24℃冰箱中保存,根据需要用甲醇水混合溶液(1∶1)稀释配制成相应浓度的混合标准工作溶液。
基质匹配标准溶液:移取10份各5 mL的空白样品提取液于15 mL样品瓶中,以真空氮气吹干仪吹至近干,分别加入1 000 μg/L的农药混合标准溶液10、20、50、100、200、500、1 000、2 000、4 000、5 000 μL于样品瓶中,各补加甲醇至5 mL,再加5 mL 8 mmol/L甲酸铵水溶液,配制成1.0、2.0、5.0、10、20、50、100、200、400、500 μg/L相应的基质匹配标准溶液。基质匹配标准工作溶液现用现配。
1.2 色谱质谱条件
1.2.1 色谱条件 色谱柱:Accucore aQ柱(100 mm×2.1 mm,2.6 μm)、Accucore C18预柱(10 mm×2.1 mm,2.6 μm)均为美国Thermo Fisher Scientific公司产品。柱温:35℃;流动相A为0.1%甲酸-4 mmol/L甲酸铵水溶液,流动相B为0.1%甲酸-4 mmol/L甲酸铵甲醇溶液。梯度洗脱程序:0~1 min,100%A;1~35 min,100% ~0%A;35~40 min,0%A;40~40.1 min,0% ~100%A;40.1~45 min,100%A;流速为300 μL/min;碰撞气:高纯氩气(纯度≥99.999%),碰撞气压力:0.2 Pa;进样量:10 μL。
1.2.2 质谱条件 电喷雾离子化,正负离子模式自动切换,鞘气压力:275 kPa,辅助气流速3 L/min,毛细管电压:正离子模式3 000 V,负离子模式2 500 V,Tube lens补偿电压:152 V,Skimmer电压:20 V。毛细管温度350℃,雾化温度(辅助气):295℃,扫描速度:0.2 s,扫描方式:动态多反应监测(EZ MRM)。质谱分析参数见表1。
1.3 样品前处理方法
分别取试样于食品捣碎机粉碎,过0.3 mm筛,混匀。称取粉碎混匀试样各10 g(精确至0.01 g)于50 mL具塞离心管中,加入20 mL水浸泡30 min,加入20 mL 1%乙酸-乙腈混合溶液、2.0 g氯化钠涡旋混合30 s后加入6.0 g无水硫酸镁、1.5 g无水醋酸钠,涡旋30 s,振荡提取5 min,以10 000 r/min离心5 min,取上层溶液10 mL于预先加有75 mg GCB、400 mg PSA及1.2 g无水硫酸镁的离心管中,高速涡旋30 s,加入0.7 mL甲苯,高速涡旋30 s,以10 000 r/min离心5 min,上层溶液经0.20 μm有机系微孔膜过滤,取200 μL滤液至进样瓶中,加入300 μL乙腈及500 μL 8 mmol/L甲酸铵溶液。供高效液相色谱-串联四极杆质谱联用仪测定。
2 结果与讨论
2.1 农药品种的筛选
根据世界各国和我国茶叶中农药的最高残留限量标准[1],本研究组对400种农药残留的检测条件进行了优化,在实验中发现:没有找到母离子(如双甲脒、DMSA)、子离子(如百草枯、四氟菊酯)和仪器响应过低(如 唑脒、拿草特)的农药有90多种,加标回收率小于70%或大于130%的农药(如 唑隆、除喹禾灵)有10种。最终确定对290种农药残留进行测定。
2.2 质谱条件的确定
用仪器自带的针泵以流动注射方式将样品通过三通与流动相混合后,分别在电喷雾离子源正离子电离(ESI+)模式和负离子电离(ESI-)模式下对290种农药的单一标准溶液进行母离子全扫描,选择母离子响应较高的模式,再对其子离子全扫描,每个化合物选择2对响应值较高的特征离子对作为定量及定性离子对进行MRM参数优化,优化的质谱参数见表1。其中双特松与敌敌畏,二丙烯草胺与咯喹酮,氯苯胺、灵敌草、净辛噻酮、西草净及辛噻酮等相对分子质量相同,保留时间一致,相互间易产生干扰,所以这些化合物需再加入1对定性离子对予以区别。
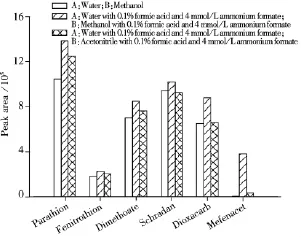
图1 不同流动相体系下6种农药的色谱峰响应值Fig.1 Chromatographic response of 6 pesticides in different mobile phases
2.3 色谱条件的优化
2.3.1 色谱柱的选择 色谱柱采用Thermo Scientific Accucore 2.6 μm填料,在加快分析速度、提高色谱柱分离度的同时,还能增加峰容量,改善复杂基质中化合物的分离,同时消除了使用小颗粒填料所造成的高反压问题。本文考察了Accucore系列中RP-MS、C18和aQ 3种不同选择性的 HPLC色谱柱(100 mm×2.1 mm,2.6 μm)对上述290种农药的分离效果。结果表明采用Accucore aQ色谱柱可获得最理想的分离效果。
2.3.2 流动相的优化 由于电喷雾质谱的电离是在溶液状态,因此流动相的组成和添加剂除了影响分析物的保留时间和峰形外,还会影响到分析物的离子化效率,从而影响到目标化合物的检测灵敏度。本实验分别以甲醇-水、乙腈-水作为流动相考察290种农药的分离度、峰形及响应值,以对硫磷(Parathion)、杀螟松(Fenitrothion)、乐果(Dimethoate)、八甲磷(Schradan)、二氧威(Dioxacarb)和苯噻草胺(Mefenacet)为例,发现以甲醇-水为流动相的离子化效率明显好于乙腈-水流动相。实验发现加入甲酸和甲酸铵可提高目标化合物的响应值(见图1),而加入乙酸铵响应值降低,因此确定以0.1%甲酸-4 mmol/L甲酸铵水溶液和0.1%甲酸-4 mmol/L甲酸铵甲醇溶液为流动相。图2为按“1.3”方法进行前处理后乌龙茶混合标准溶液的总离子流色谱图。
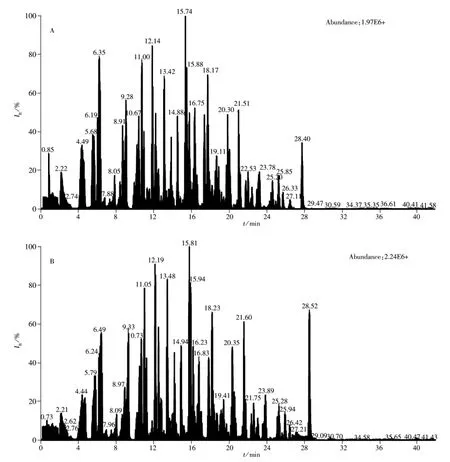
图2 290种农药的乌龙茶标准溶液(20 μg/L)的总离子流色谱图Fig.2 Total ion current chromatograms of 290 pesticides standard at 20 μg/L prepared in oolong tea
2.4 样品前处理条件的优化
2.4.1 提取条件的优化 茶叶为干性样品,加入乙腈提取前用水浸泡30 min,可以增加溶剂的提取效率。加入氯化钠使得水与乙腈分层,涡旋后再加入无水硫酸镁,其中氯化钠需先于无水硫酸镁加入溶液,避免同时加入后无水硫酸钠遇水直接结块,从而将目标化合物包裹,影响农药的提取率。无水硫酸镁用于吸取盐析分配后有机层中多余的水分。为研究无水硫酸镁及无水硫酸钠的使用对于农药提取效果的影响,按照“1.3”的前处理方式分别加入6.0 g无水硫酸镁与6.0 g无水硫酸钠,农药添加水平为10 μg/L,以苯噻草胺(Mefenacet)、灭线磷(Ethoprophos)、咪唑菌酮(Fenamidone)、苄草隆(Cumyluron)、杀草净(Dipropetryn)和嘧菌酯(Azoxystrobin)6种农药的回收率为考察对象。其中苯噻草胺是除草剂中的噻唑多环化合物,灭线磷是有机磷类杀虫剂,咪唑菌酮为含苯胺基的杀菌剂,苄草隆为含苯甲基除草剂,杀草净为三嗪类芽前除草剂,嘧菌酯为甲氧基丙烯酸酯类杀菌剂,这些化合物能代表大多数化合物的结构特点。结果发现使用无水硫酸镁的回收率优于无水硫酸钠(见图3),原因为在净化步骤中采用的PSA属于正相吸附剂,含水量越少净化效果越好,无水硫酸镁的吸水能力更佳,且其粒度更小,在振摇与涡旋的过程中可使样品更加粉碎,并通过与乙腈发生协同作用,提高提取效率[18]。
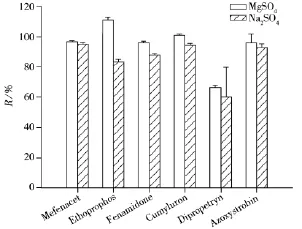
图3 2种QuEChERS样品前处理方法对10 μg/L加标乌龙茶样品的回收率比较(n=7)Fig.3 Comparison of recoveries of two versions of QuEChERS sample preparation method for oolong tea samples spiked with 10 μg/L pesticides(n=7)
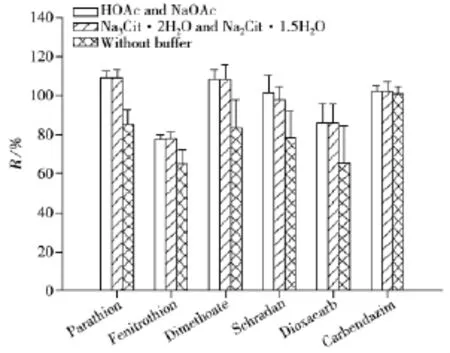
图4 3种QuEChERS样品前处理方法对10 μg/L加标乌龙茶样品的回收率比较(n=7)Fig.4 Comparison of recoveries of three versions of QuEChERS sample preparation method for oolong tea samples spiked with 10 μg/L pesticides(n=7)
由于涉及的农药种类较多,为了保证酸碱不稳定的农药不分解,需要加入缓冲盐体系来保持基质环境的pH值。因此,考察了缓冲盐的使用对于农药提取效果的影响,根据2003 Oringinal、国际官方方法AOAC 2007.01与欧洲标准化委员会的标准方法CEN 151662进行实验,第一组加入1.5 g乙酸钠和在乙腈中加入1%乙酸(AOAC 2007.01),第二组加入0.5 g倍半水合柠檬酸二钠和1.0 g二水合柠檬酸钠(CEN 151662),第三组不加缓冲盐(Oringinal)。农药添加水平为10 μg/L,以对硫磷(Parathion)、杀螟松 (Fenitrothion)、乐果(Dimethoate)、八甲磷(Schradan)、二氧威(Dioxacarb)和多菌灵(Carbendazim)6种农药的回收率为考察对象。结果发现加入乙酸和乙酸钠后,除多菌灵的回收率变化不大,对硫磷、杀螟松、乐果、八甲磷和二氧威的回收率均有所提高(见图4)。其原因为采用乙酸和乙酸钠体系可保证样品基质环境的pH值为5.0~5.5,这既可以保证碱不稳定的农药(如对硫磷、杀螟松、乐果)的回收率,也可以保证酸不稳定的农药(如乐果、八甲磷)的回收率,而多菌灵在酸碱溶液中均具有良好的稳定性,所以其回收率变化不大。第一组与第二组均可保证样品基质的pH值在整个过程中分别保持在5.0~5.5与4.8,且以上6种农药的回收率十分接近,但从实验缓冲能力与成本考虑,最终选择乙酸与乙酸钠缓冲盐体系。
2.4.2 净化条件的优化 Oringinal、国际官方方法AOAC 2007.01与欧盟标准方法CEN 151662的另一处不同在于前两者均未提及添加C18与石墨化炭黑(GCB),而后者可以。C18属于反相吸附剂,可有效除去基质中的脂肪和脂类等非极性有机物,为研究C18的使用对于农药提取效果的影响,分别进行添加500 mg C18和不添加C18对比实验,农药添加水平为10 μg/L,以苯噻草胺(Mefenacet)、灭线磷(Ethoprophos)、咪唑菌酮(Fenamidone)、苄草隆(Cumyluron)、杀草净(Dipropetryn)和嘧菌酯(Azoxystrobin)6种农药的回收率为考察对象。结果发现两组农药的回收率相近(见图5),其原因为茶叶中的脂类等非极性有机物含量较低,而且在提取溶剂中溶解较少。所以针对茶叶样品,本实验不加入C18。
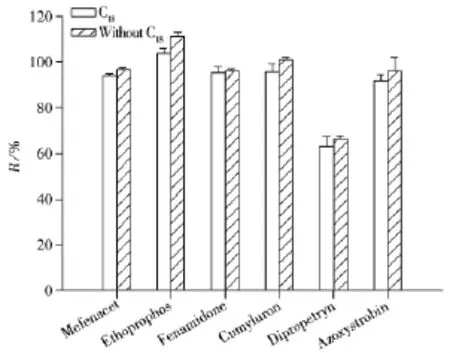
图5 2种QuEChERS样品前处理方法对10 μg/L加标乌龙茶样品的回收率比较(n=7)Fig.5 Comparison of recoveries of two versions of QuEChERS sample preparation method for oolong tea samples spiked with 10 μg/L pesticides(n=7)
欧盟标准方法CEN 151662规定,在深色样品中可加入少量石墨化炭黑(GCB)除色,茶叶样品经溶剂提取后(不加入GCB),由于其提取液中存在叶绿素、胡萝卜素、叶黄素与花青素,且PSA只能除去少量色素,提取液呈墨绿色(绿茶)、棕红色(乌龙茶、红茶)、棕黑色(普洱茶)。为了确定GCB用量,分别在10 mL各种茶叶提取液中添加40、45、50、55、60、65、70、75、80、85 mg GCB,高速涡旋30 s,以10 000 r/min离心5 min,观察提取液颜色。实验发现,当加入75 mg GCB时,提取液为黄色透明(乌龙茶、红茶、普洱茶)或无色透明(绿茶)。但GCB外层为6个碳原子构成的平面六角形,对于片状化合物有强烈的吸附作用,随着GCB的加入会导致片状农药(如灭线磷 (Ethoprophos)、氧环唑(Azaconazole)、腈苯唑(Fenbuconazole)、吡螨胺(Tebufenpyrad)、克百威(Carbofuran)、嘧霉胺(Pyrimethanil)的回收率降低,通过在提取液中加入甲苯可以洗脱被GCB吸附的农药,提高该类农药的回收率。为了确定甲苯的加入量,实验中加入75 mg GCB,农药添加水平为10 μg/L,以上述6种农药的回收率为考察对象,结果见图6,发现加入0.7 mL甲苯可将被GCB吸附的农药洗脱;但会对原来未加入甲苯时回收率好的农药,例如敌敌畏(Dichlorvos)、非草隆(Fenuron)造成影响,原因为溶液中基质杂质增加。综合上述结果,实验条件确定为取上层溶液10 mL于预先加有75 mg GCB、400 mg PSA及1.2 g无水硫酸镁的离心管中,高速涡旋30 s,再加入0.7 mL甲苯。

图6 甲苯加入量对部分农药回收率的影响Fig.6 Influence of toluene volume used in purification on recoveries of 8 pesticides
根据2003 Oringinal、国际官方方法AOAC 2007.01与欧盟标准方法CEN 151662中各种提取剂与净化剂的用量,本实验在整个过程中对PSA、无水硫酸镁、氯化钠与缓冲盐的用量进行了优化,过程与上述方法相似,形成了适合于茶叶样品的QuEChERS前处理方式(见“1.3”)。
2.4.3 基质效应 目标化合物的电离容易受到基质的干扰,导致离子抑制或增强,基质匹配标准溶液的响应值比纯溶剂的高(见图2),且这种影响在各待测物之间存在差异,表明不同农药受到基质的影响不同。当基质效应影响过大时,会降低方法的灵敏度,影响方法的准确性,因此需要对基质效应进行评价。评价方法为:基质效应=(1-基质匹配标准曲线的斜率/纯溶剂标准曲线的斜率)×100%[19]。结果表明在290种农药中普遍存在基质效应,其中19.2%属于中等强度的基质效应,2.5%属于强基质效应,79.3%为较弱基质效应。为最大限度消除基质效应干扰,本实验采用基质匹配法进行定量分析。以空白茶叶样品的提取液为标准溶液的稀释溶液,可使标准溶液和样品溶液具有相同的离子化环境,从而消除基质效应。
由于测定的化合物较多,本实验对化合物保留时间的稳定性进行了研究,分别对质量浓度为20 μg/L的混合标准溶液连续测定7次,同样方法配制标准溶液的化合物出峰时间稳定,漂移时间均小于1.2 s;基质匹配的标准溶液较纯标准溶液出峰晚1~9 s,这与María等[20]的测定结果相同,其原因为基质进入色谱后影响了色谱柱的柱效。
2.5 方法学考察
2.5.1 标准曲线与定量下限 配制290种农药质量浓度依次为1.0、2.0、5.0、10、20、50、100、200 μg/L的系列混合标准溶液。以目标组分的峰面积(y)对相应的质量浓度(x,μg/L)绘制标准曲线。结果发现,其相关系数(r2)均大于0.99,各种农药质量浓度在1.0~200 μg/L范围内具有较好的线性关系。用本方法对绿茶、红茶、乌龙茶、普洱茶样品进行加标回收实验,以信噪比(S/N)≥10确定290种农药的定量下限均可达到0.01 mg/kg。
2.5.2 精密度与加标回收率 对比CAC、欧盟、日本、中国茶叶农残限量标准规定,欧盟的最高残留限量(MRL)的规定最为严格,本文根据欧盟最高残留限量(没有MRL规定的按照日本《食品中农业化学品肯定列表制度》中的“一律标准”与欧盟规定的0.01 mg/kg标准)的1倍、2倍、4倍进行加标回收实验,结果表明目标分析物的回收率均在61%~119%范围内,重复实验7次,其相对标准偏差小于12.4%,加标浓度为MRL(没有限量规定的按照0.01 mg/kg进行加标)的相对标准偏差和平均回收率测定结果见表1。

表1 290种农药的保留时间、质谱分析参数、相关系数(r2)、平均回收率与精密度(n=7)Table 1 Retention times,mass spectrometric parameters,correlation coefficients(r2),recoveries and precisions of 290 pesticides(n=7)
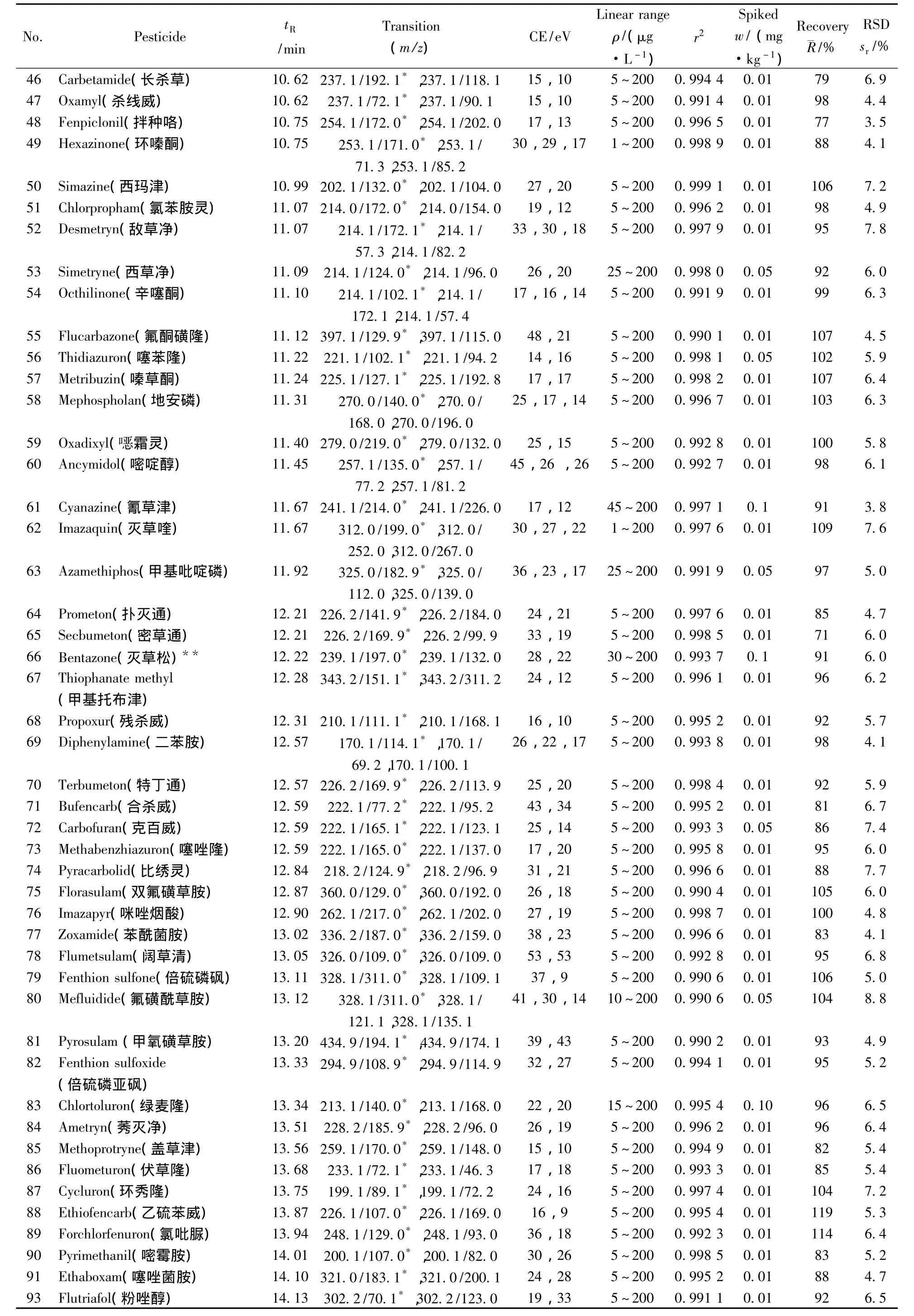
(续表1)
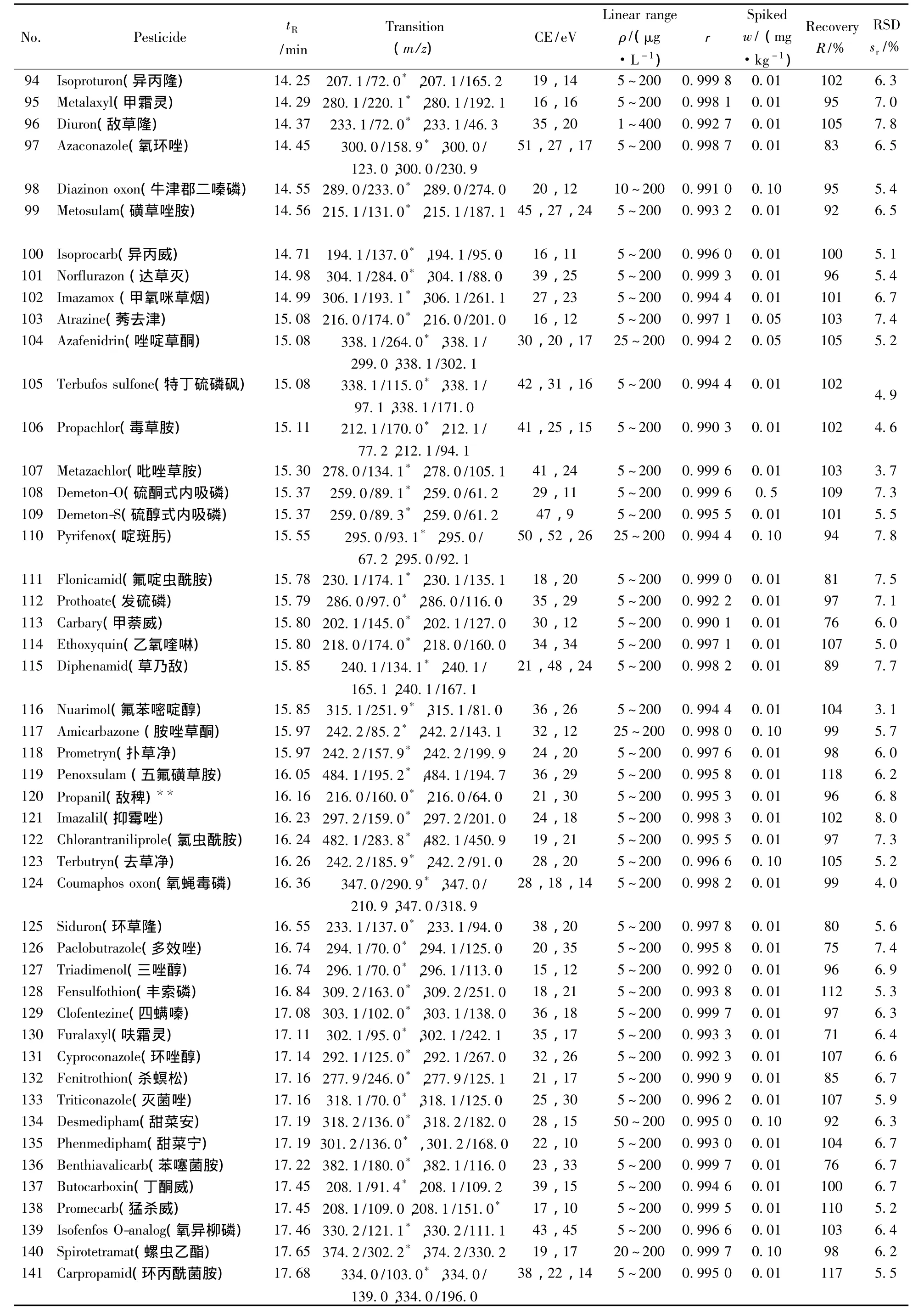
(续表1)
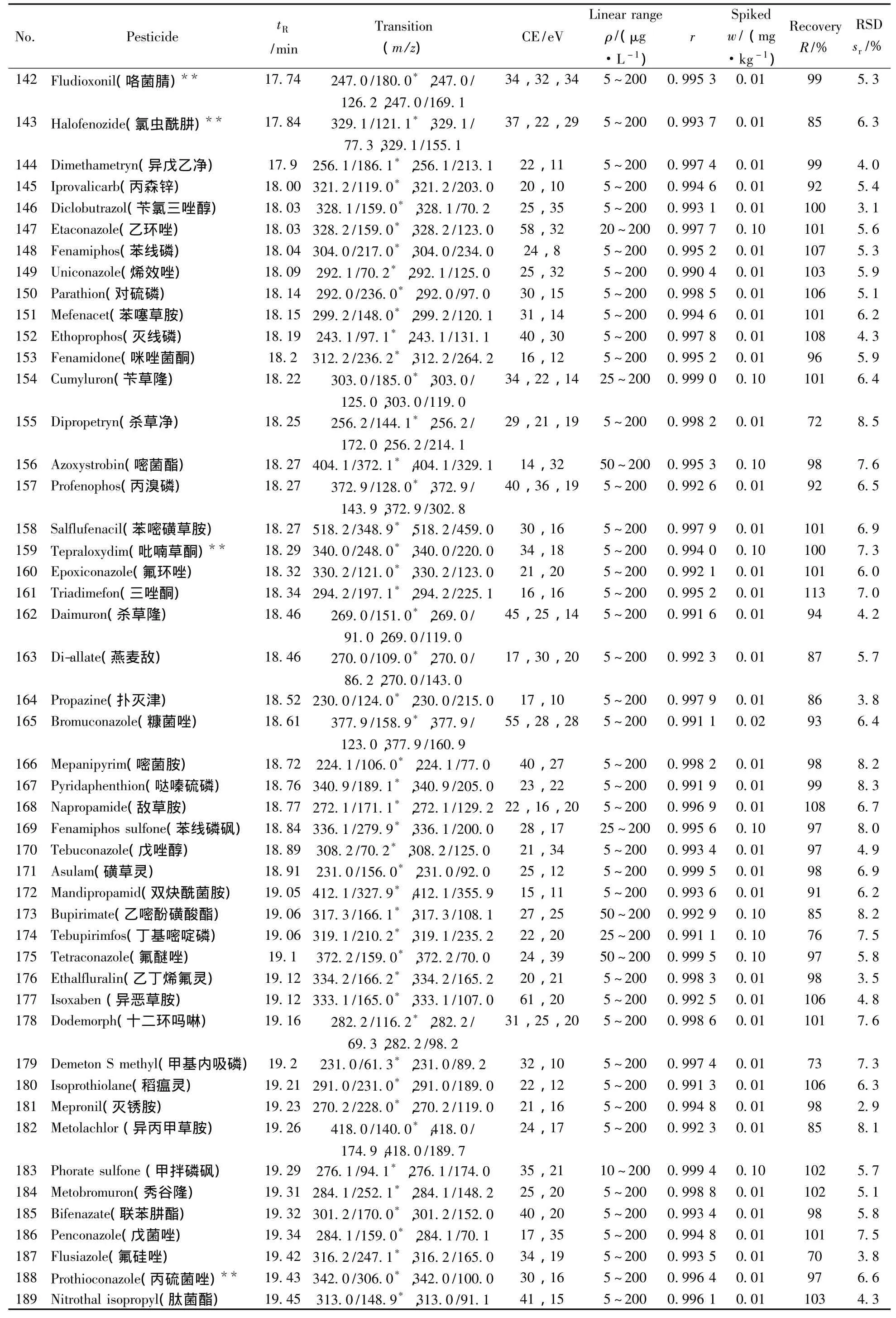
(续表1)
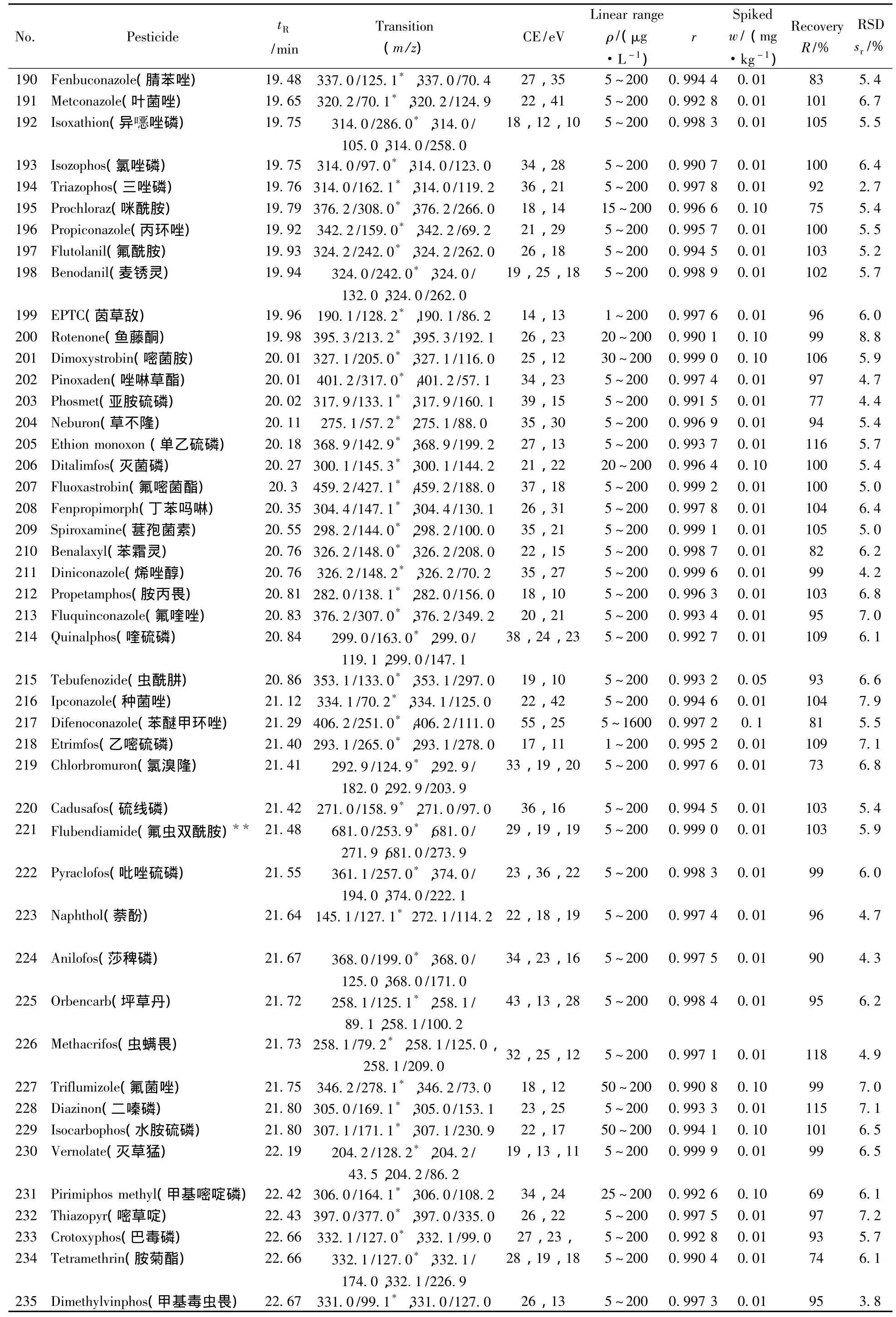
(续表1)
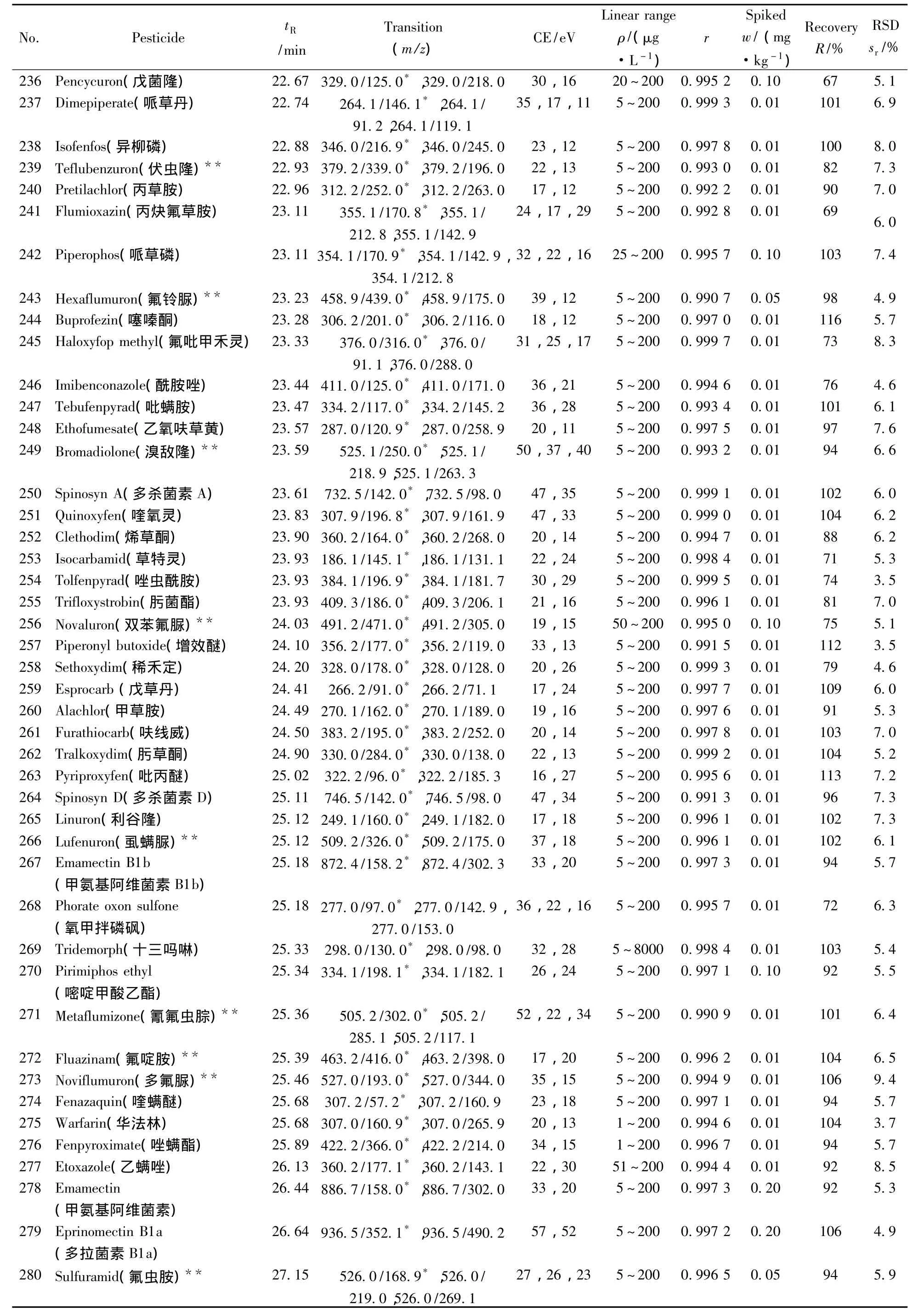
(续表1)
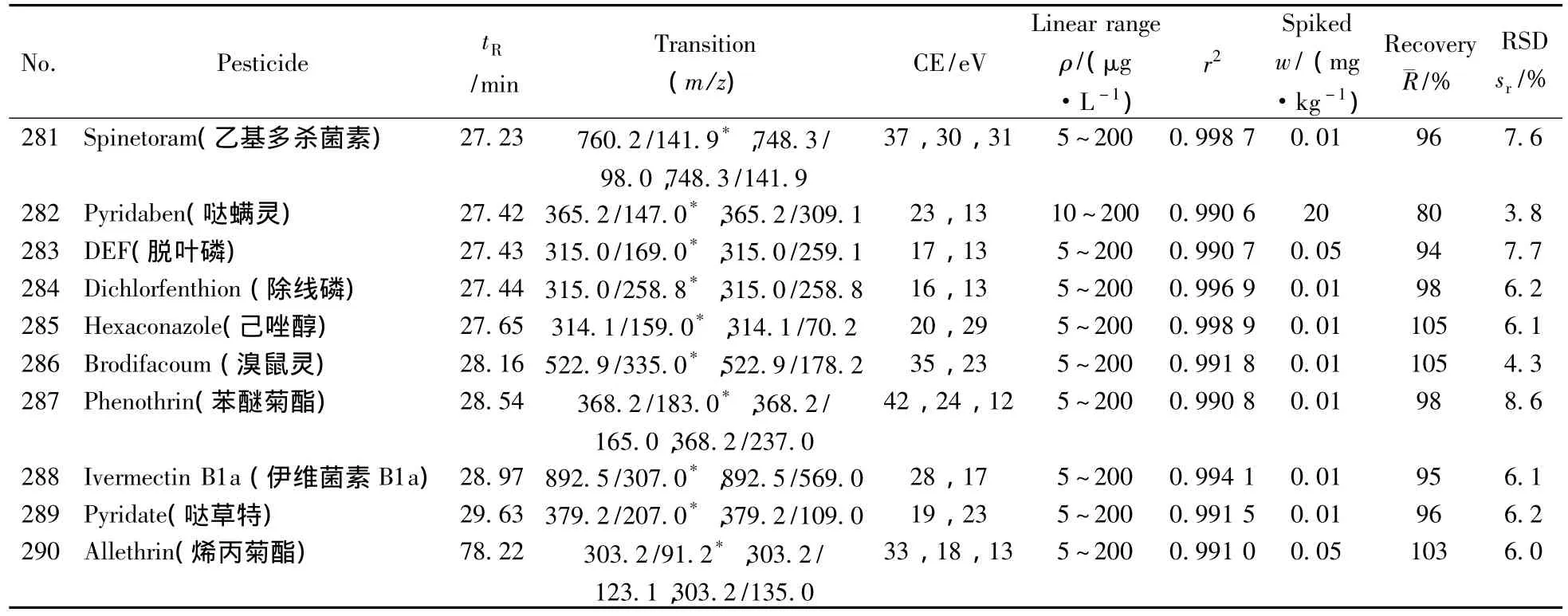
(续表1)
2.6 实际样品分析
采用本方法对市售绿茶、乌龙茶、红茶、普洱茶样品共10份进行了290种农残检测,其中1份检出啶虫脒(Acetamiprid)和甲胺磷(Methamidophos),含量分别为28 μg/kg和37 μg/kg;1份检出吡虫啉(Imidacloprid)和噻虫嗪(Thiamethoxam),含量分别为12 μg/kg和19 μg/kg;1份检出乙酰甲胺磷(Acephate)、乐果(Dimethoate)、氧化乐果(Omethoate)、三唑磷(Triazophos)、亚胺硫磷(Phosmet)、三唑酮(Triadimefon)、噻嗪酮(Buprofezin)、水胺硫磷(Isocarbophos)、喹硫磷(Quinalphos)、哒螨灵(Pyridaben)和吡虫啉(Imidacloprid),含量分别为 18、26、39、43、29、68、82、29、46、25、14 μg/kg。研究结果表明,本方法可用于茶叶中290种农药残留的检测。
3 结论
本文利用高效液相色谱-串联质谱联用技术(HPLC-MS/MS)进行测定,采用QuEChERS方法对样品进行前处理,建立了茶叶中290种农药残留量的测定方法,方法具有良好的灵敏度、准确度和精密度,线性关系良好,能满足我国、日本和欧盟等国家与组织对于多农残分析的要求。
致谢:感谢美国Thermo Fisher Scientific公司James Chang和Jia Wang工程师在实验过程中的帮助和支持。
[1] General Administration of Quality Supervision,Inspection and Quarantine of the People s Republic of China.Pesticide Residue Limits(国家质量监督检验检疫总局.农药残留限量查询).[2012-06-17].http://www.tbtsps.com/FOODSAFE/XLBZ/Pages/pesticide.aspx.
[2] Nuria S,Bego a V,Berta B,Marco M.Talanta,2012,89(30):310 -316.
[3] Flaviane A S,Anna I G C,Maria E L R Q,Reinaldo F T,Ant nio A N,Gevany P P.Food Chem.,2012,135(1):179-185.
[4] Liu D,Min S G.J.Chromatogr.A,2012,1235(27):166-173.
[5] Chen L,ShangGuan L M,Wu Y Y,Xu L J,Fu F F.Food Control,2012,25(2):433 -440.
[6] Li B,Zeng F G,Dong Q C,Cao Y,Fan H T,Deng C F.Phys.Procedia,2012,25(3):1776 -1780.
[7] Milagros M,Angeles M,Amadeo R.J.Chromatogr.,2012,1100(3):605-619.
[8] Virginia P,Elena D,Bezhan C,Antonio L,Maria G,Maria L M.J.Chromatogr.A,2012,1234(20):22-31.
[9] Claudia O,Wolfgang S.J.Chromatogr.A,2011,1218(37):6540-6547.
[10] Emilia F,Anna S.J.Chromatogr.B,2012,901(15):107-114.
[11] Oscar N,Héctor G,Imma F,Encarnación M,Maria T G.J.Chromatogr.A,2012,1249(3):164 -180.
[12] László P,Juan F G R,Péter F,Attila G,Mihály D,László A,Bienvenida G,Antonio M.J.Chromatogr.A,2012,1249(3):83-91.
[13] Zhu W X,Yang J Z,Liu Y F,Wei W.J.Instrum.Anal.(祝伟霞,杨冀州,刘亚风,魏蔚.分析测试学报),2010,29(11):1109-1113.
[14] Xu J,Chen J,Ye H Y,Wang L,Sun L H,Lai Z F.J.Instrum.Anal.(徐娟,陈捷,叶弘毅,王岚,孙灵慧,赖子峰.分析测试学报),2011,30(9):990-995.
[15] Anastassiades M,Lehotay S J,Stajnbaher D,Schenck F J.J.AOAC Int.,2003,86(2):412 -431.
[16] Lehotay S J,Katerˇina M,Alan R L.J.AOAC Int.,2005,88(2):615 -629.
[17] Anastassiades M.prEN 15662.Determination of Pesticide Residues Using GC-MS and/or LC-MS(/MS)Following Acetonitrile Extraction/Ppartitioning and Ccleanup by Dispersive SPE-QuEChERS Method.European Commitee for Standardization.
[18] Lehotay S J.J.AOAC Int.,2007,90(2):485 -520.
[19] Anastasios E,Helen B,Spyros A,Despina T.J.Chromatogr.A,2009,1216(31):5856 -5867.
[20] María L G P,Patricia P B,Roberto R G,José L M V,Antonia G F.J.Chromatogr.A,2012,1248(27):130-138.
猜你喜欢
中国土壤与肥料(2021年5期)2021-12-02
今日农业(2020年22期)2020-12-14
化工设计通讯(2020年10期)2020-09-17
中国蜂业(2018年4期)2018-05-09
Cancer Biology & Medicine(2016年4期)2017-01-13
中国氯碱(2016年9期)2016-11-16
当代化工研究(2016年6期)2016-03-20
电力需求侧管理(2014年4期)2014-03-20
无机化学学报(2014年3期)2014-02-28
河南科技(2014年12期)2014-02-27
