
运用近邻传播聚类分析进行SELDI-TOF 蛋白质谱特征选择
2013-03-10 08:11杨合龙厉力华郑智国孟旭莉杭州电子科技大学生命信息与仪器工程学院杭州3008
中国生物医学工程学报 2013年1期
杨合龙 祝 磊 韩 斌 厉力华* 郑智国 孟旭莉(杭州电子科技大学生命信息与仪器工程学院,杭州 3008)
2(浙江省肿瘤研究所,杭州 310022)
引言
随着高通量蛋白质组学的发展,基于蛋白质组学的质谱技术可以使癌症在发生的早期阶段被发现和及时治疗,从而大大提高了癌症患者的治愈率[1]。研究表明,在众多蛋白质中,只有少数与某种特定疾病有关,这些少数特定的蛋白质被称为生物标志物(biomarker)[2]。近年来,研究者利用各种方法来寻找这些生物标志物[3]。表面增强激光解析电离飞行时间质谱(surface enhanced laser desorption/ ionization time-of-flight mass spectrometry,SELDI-TOF MS)技术是目前最为可行的蛋白组学技术之一。一份SELDI-TOF 样本数据的特征维数可达上万维,而样本数量一般仅为几十到上百例,这从模式识别的角度被称为“维数灾难”[4]。因此,如何有效地降低特征维数,去除不相关的蛋白质位点,进而筛选出生物标识物,是蛋白质组学的一个热点问题[5]。
聚类分析是一种通过计算数据之间的相似性来将数据分类的技术,已经在基因微阵列数据分析中得到了成功应用,对于降低特征维数和去除特征间相关性效果显著。Hancer 等利用K-means 聚类算法对原始的基因微阵列数据集进行聚类,然后计算每一个类中的平均强度作为“原型基因”参加最后的样本分类过程,但不是所有的“原型基因”都与样本分类相关,因此必然会引入冗余从而影响分类结果[6]。Wang 等利用分级聚类算法进行聚类,数据形成一个树枝状图,通过在不同强度下剪切树枝状图,将数据分成不同的类,在每一个类中选出一个最有代表性的数据,但是算法的复杂性较高,运算强度较大[7]。对于蛋白质质谱数据,Yang 等设计一个迭代算法,通过30 次的迭代来克服K-means 聚类算法的随机性和不稳定性[8],之后再利用遗传算法(GA)[9]来提取生物标志物,迭代算法的计算强度较大,虽然已进行了迭代,但算法中仍存在不稳定因素。
为得到较好的分类结果,避免算法的随机性和不稳定性,本研究提出一种基于近邻传播聚类分析的特征选择方法。首先,通过t-test 对数据进行初筛;然后,运用近邻传播聚类分析和零空间LDA(null space linear discriminant analysis)[10],对数据进行降维,同时降低特征间的相关性;最后,利用
SVM-RFE(support vector machine recursive feature elimination)进行特征选择,挑出最具有判别意义的蛋白质位点。在卵巢癌公共数据集OC-WCX2a、OC-WCX2b 和浙江省肿瘤医院乳腺癌临床数据集BC-WCX2a 上的实验结果表明,该方法不但能降低数据维数,获得较高的分类率,而且还确定了具有判别意义的蛋白位点,有助于指导后续的蛋白生化鉴定,为癌症发病机制的研究提供了参考。
1 方法与数据
1.1 方法简介
1.1.1 近邻传播聚类分析
在数据挖掘和数据分析中,聚类分析已经得到了广泛的应用。对于基于蛋白质组学的质谱数据,其数据量是巨大的,应用聚类分析主要在于去除多余的冗余,以达到减少数据的目的。通过对质谱数据进行聚类,得到特定数目的类,类内数据相似度最强,类间数据相似度最弱,每一个类中选择特定的“代表”来代替整个类。应当看出,通过聚类分析并不是武断地删除数据,而是通过分析数据之间的特性,从中选出特定“代表”来代替部分数据,从而达到减少数据的目的。
近邻传播聚类分析 (affinity propagation clustering,AP)是一种基于近邻信息传播的聚类算法[11],目的是寻找最优的类代表集合,使所有样本达到最近的类代表的相似度之和最大,其主要特点是适合处理大规模数据。与K-means 聚类算法相比,AP 算法本身克服了随机性,因此较为稳定。
AP 算法将所有数据点都看作是潜在的类代表点,首先建立相似矩阵s,此相似矩阵s(i,k)= -xi-xk2 表示任意两个数据点xi和xk之间的负欧几里得距离。在聚类之前,初始假定所有的样本被选为类代表的可能性相同,即设定所有的s(k,k)为相同的值p。p 值为偏向参数,表示数据点选择自己作为聚类中心的意愿,其值越大,聚类结果的类数目越多,每个类中所含的成员数目就越少。
该聚类过程是对数据点不断提取信息同时又不断进行处理,在聚类过程中引入两个参数:可信度[responsibility,记作r(i,k)]和可用度[availability,记作a(i,k)]。聚类的核心步骤就是这两个参数交替更新的过程。参数可用度(availability)初始化为0,即a(i,k)= 0。
a(i,k)表示数据点i 被选为成为数据点k 的类中心的积累过程,有
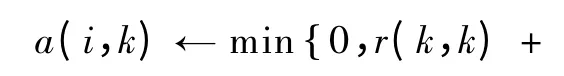
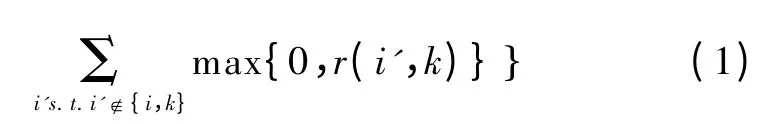
式中,r(i,k)表示数据点k 被选为数据点i 的类中心的积累过程,有
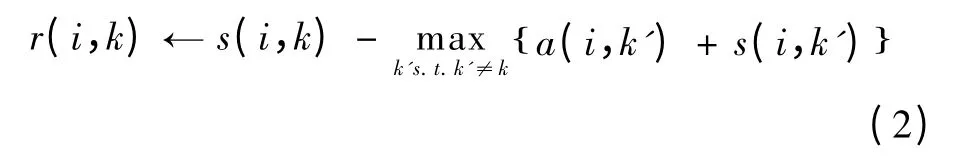
对于任何点来说,要确定其聚类中心,可信度(responsibility)和可用度(availability)都是必须考虑的。对于数据点i,使得式(3)最大的数据点k 可以作为数据点i 的类中心点,有
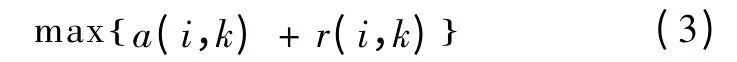
1.1.2 零空间LDA 算法
对于聚类之后的SELDI 数据,特征维数仍远大于样本个数。若直接使用经典 LDA (linear discriminant analysis)算法,将会导致类内散布矩阵SW奇异。类内散布矩阵SW、类间散布矩阵SB和总体散布矩阵ST以及最优投影方向Wopt可计算如下:
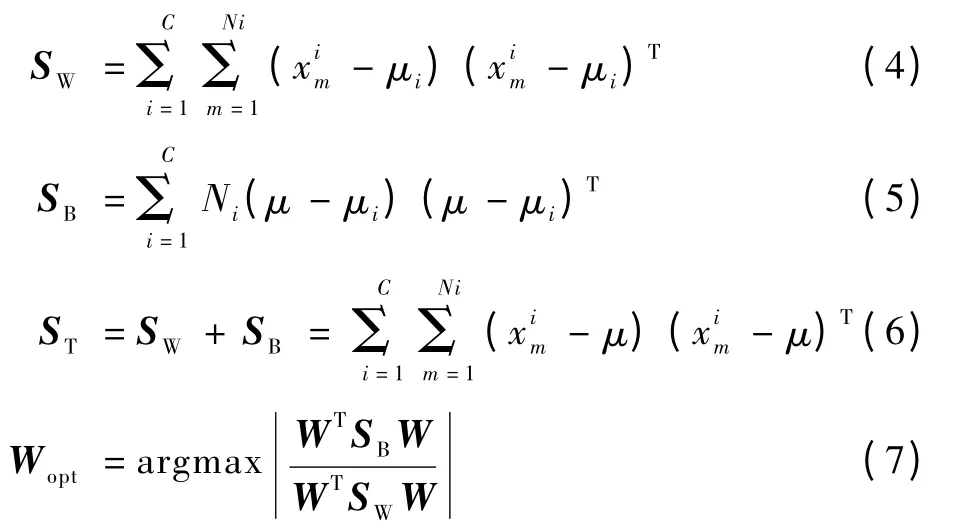
零空间LDA 首先去除ST的零空间,这样既去除了SB和SW交叉的零空间部分,又保留了判别信息,之后在SW的零空间上找到使SB最大的方向(即为最佳的投影方向)的Wopt。
1.1.3 支持向量机递归特征去除算法
经过AP 聚类算法和零空间LDA 算法处理之后,SELDI 数据特征之间的相关性已大大减少,然后利用支持向量机递归特征去除算法(support vector machine recursive feature elimination,SVM-RFE)进行特征选择,找出最具有判别意义的特征。
SVM 首先通过非线性变换,将输入空间变换到一个高维特征空间,然后再在这个新空间中求取最优线性分类面,其最优分类面可以最大化分类间隔(Margin)M = 2/‖w‖,等价于使‖w‖2最小。SVM-RFE 方法在进行特征选择时,首先训练SVM分类器,然后按标准计算其特征的排列,再移去排列最后的特征。不断循环,直到标准没有明显减小或者已经得到需要的特征数为止[12]。
SVM-RFE 特征选择算法过程如下:在每一次循环中,具有最小排列系数的特征将被移除,然后SVM 对余下的特征重新训练,以获取新的排序系数。SVM-RFE 方法通过迭代执行这一过程,最后得到一个特征排序表。利用该排序列表,定义若干个嵌套的特征子集来训练SVM,并以SVM 的预测正确率来评估这些子集的优劣,从而获得最优的特征子集。需要注意的是,排在前面的那些特征,单个并不一定能使SVM 分类器获得最好的分类性能,而是多个特征组合在一起,才能使分类器获得最优的分类性能。因此,SVM-RFE 算法能选择出互补的特征组合。
1.1.4 皮尔逊相关系数
利用皮尔逊相关系数,对所选特征之间的相关系数进行计算。计算筛选出的特征之间的平均相关系数为
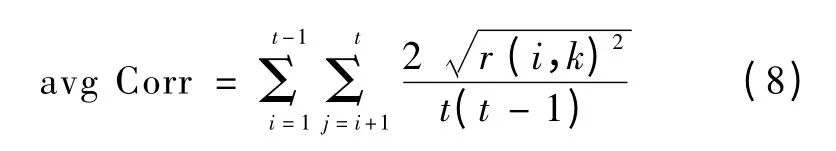
式中,t 为所选出的特征数,r(j,k)为一对特征(第j个特征和第k 个特征)的皮尔逊相关系数。
r(j,k)可由下式计算得到,即
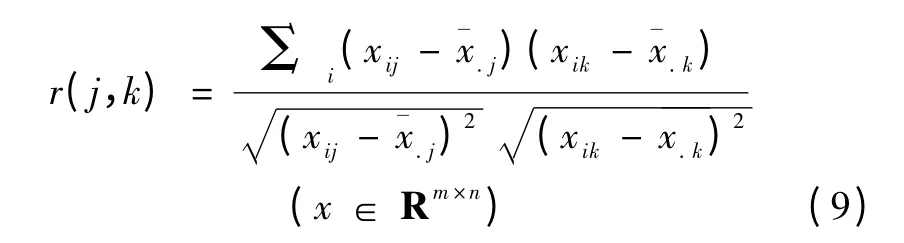
1.2 数据
为了验证所提方法的有效性,分别在3 个临床蛋白质谱数据集上进行实验。
1.2.1 卵巢癌公共数据集 OC-WCX2a 和 OCWCX2b
数据由美国食品和药物管理局(FDA)及国家肿瘤研究所(NCI)提供。采用WCX2 蛋白芯片阵列和SELDI-TOF 技术,对人体血清样本进行分析。数据集包含两类蛋白表达信息:癌症患者(cancer)和正常对照组(control)。其中:OC-WCX2a,Cancer 100 例,Control 100 例;OC-WCX2b,Cancer 162 例,Control 91 例。每个质谱样本包含15 154 维质核比值(m/z)/强度值(Intensity)特征。上述两个数据集可以在http://home.ccr.cancer.gov/下载,并且包含有数据集的详细介绍。
1.2.2 乳腺癌临床数据集BC-WCX2a
该数据由浙江省肿瘤医院提供,共包含126 例血清蛋白质质谱样本,其中Cancer 87 例,Control 39例,每例样本约为65 536维蛋白特征[13]。
1.3 数据预处理方法
虽然蛋白质组学质谱技术产生的数据包含大量的潜在信息,但由于样本采集和质谱仪的性能,使数据中含有大量噪声,对蛋白质组学质谱数据分析提出挑战,因此在质谱数据分析之前需要采用数据预处理步骤。
步骤1:重采样。由医学知识得知,蛋白质特征表述区间一般为1 500 ~10 000 Da,且质谱仪的优化区间段也是1 500 ~10 000 Da,因此选择此区间范围内的数据进行均匀重采样,采样后得到10 000个特征点。
步骤2:基线去除。基线通常位于低质荷比区域,由样本或矩阵分子中的污染引起,可以看作是显著峰与噪声间的边界[14]。由于谱的偏移程度不同,通常将每个谱的基线调整到水平线上。
步骤3:谱峰矫正。在不同的实验中,由于受到实验设备和人为因素的干扰,可能导致m/z 值出现偏差,因此对于不同批次下采集的质谱数据需要进行校准。
步骤4:归一化。使不同的质谱可以在同一个范围内进行分析,按下式归一化,有
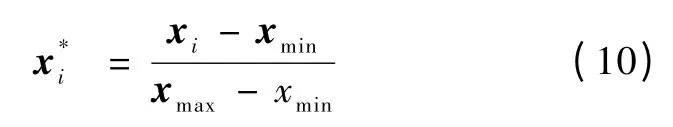
针对不同的数据集,采用不同的预处理步骤。公共数据集OC-WCX2a 和OC-WCX2b 已经过重采样、去基线、谱峰矫正等预处理,只需再进行归一化处理。乳腺癌临床数据集BC-WCX2a 经过重采样、去 除 基 线,在 4 091.1(m/z)、5 908.0(m/z)、6 634.0(m/z)等3 个基准点进行校准,最后进行归一化。
1.4 实验流程
具体实验流程如图1 所示。原始SELDI 蛋白质谱数据经过预处理后,按照十折交叉验证策略,随机分为训练集和测试集;实验共运行20 次,包括6个步骤。
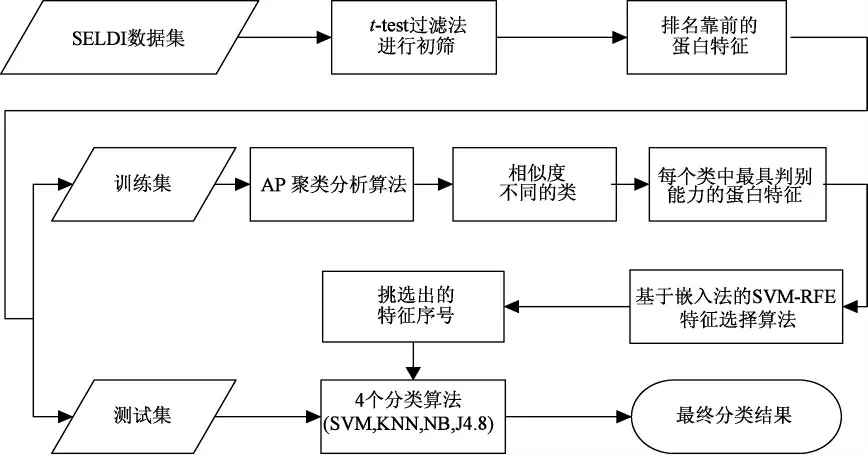
图1 基于近邻传播聚类算法的实验流程Fig.1 Flow chart of the experimentation based AP clustering
步骤1:利用t-test 对原始质谱数据进行初步筛选。为减小后续计算的复杂度,寻找潜在的生物标志物,计算每个特征的t 值并进行排序,筛选出排名靠前的2 000个特征。
步骤2:使用十折交叉验证策略,将经过初步筛选的数据分成训练集(train set)和测试集(test set)。
步骤3:对训练集数据,按照本文1.1.1 节所述AP 聚类算法对特征进行聚类。实验发现,对于OCWCX2a 数据集,当偏向参数p(preference)等于-3时,分类率等实验结果参数最优;对于OC-WCX2b数据集,偏向参数p = -30时实验结果最优;而对于BC-WCX2a 数据集,偏向参数p 取相似矩阵s 的中位值时,实验结果最优。
步骤4:由于每一类中包含很多相似性较大的特征,具有较强的相关性,所以运用零空间LDA 算法去除每一个类中一半的冗余特征,保留具有代表性的另一半特征。
步骤5:基于嵌入法的SVM-RFE 特征选择算法,通过选择有用的特征并且去除无用的特征,进一步使特征之间的关联性和冗余度最小化。
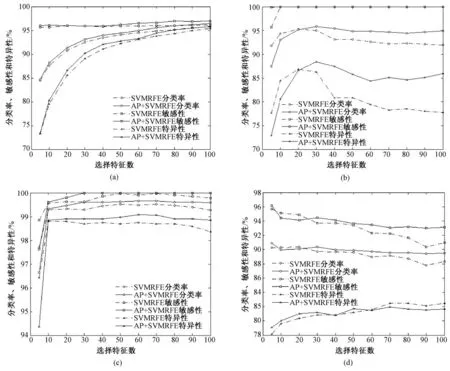
图2 3 个数据集在4 个分类器中得到的结果。(a)OCWCX2a 在SVM 得到的结果;(b)OCWCX2b 在SVM得到的结果;(c)OCWCX2b 在KNN 得到的结果;(d)BCWCX2a 在J4.8 得到的结果Fig.2 The result from three dataset in four classifiers. (a)The result of OCWCX2a in SVM;(b)The result of OCWCX2b in SVM;(c)The result of OCWCX2b in KNN;(d)The result of BCWCX2a in J4.8
步骤6:将挑选出的特征放入到4 个分类器进行验证,得到分类结果。
2 结果
2.1 分类性能
OC-WCX2a 数据上的实验结果显示,在SVM 分类器中,SVM-RFE 算法达到较好的分类效果(84.48% ~95.75 %)。在加入AP 聚类算法去相关之后,AP +SVM-RFE 算法在各特征数下的分类率都有所提高(84.70 % ~96.43 %)。因此,AP + SVM-RFE无论是在分类率、敏感性、特异性都得到了提升,最高分别达到了96.43 %、97.00 %、95.85 %。
OC-WCX2b 数据上的实验结果显示,在SVM 分类器中对比SVM-RFE 和AP +SVM-RFE 算法性能,随着特征数的增加,后者的分类率和特异性均有较大的提升,分别达到了98.99 %和97.19 %,但敏感性无较大的区别,均为100 %。在KNN 分类器中,AP + SVM-RFE 算法性能优势明显,在各特征数下,分类率、敏感性、特异性均高于SVM-RFE 算法,分别达到了99.66 %、100 %、99.08 %。虽然两图结果有差异,原因是使用了不同的分类器所造成的,但是结果显示,即使分类器不同,AP + SVM-RFE 算法性能均要高于SVM-RFE 算法性能。
BC-WCX2a 数据上的实验结果显示,在J4.8 分类器中随着特征数增加,虽然AP + SVM-RFE 在特异性方面不如SVM-RFE 算法,且分类率和敏感性有所下降,但也分别达到90.88 %和96.17 %。原因可能是由于BC-WCX2a 数据由浙江省肿瘤医院的质谱仪产生,获得时极容易被外界以及实验仪器所污染,虽然经过数据预处理,但是与公共数据相比,含有更多的噪声和影响因素,因而在特异性方面与公共数据相比不理想。
2.2 特征相关性比较
利用皮尔逊相关系数,对所选特征之间的相关系数进行计算,并比较AP + SVM-RFE 和SVM-RFE对于减少特征相关性的优劣性。
表1 为特征之间的平均相关系数,特征间平均相关系数越小,代表特征间的相关性越小,反之越大。
表1 显示,随着特征数的增加(从5 增至40),特征之间的相关系数也随之增加;但AP +SVM-RFE与SVM-RFE 相比,由于平均相关系数更小,因此AP+SVM-RFE 算法对于减少特征相关性更为有效。对比平均相关系数和分类率结果可以发现,平均相关系数与分类率之间存在关联性,AP + SVM-RFE得到的平均相关系数更低,因此筛选出的低相关性特征得到的分类率更高。分析原因,是因为AP +SVM-RFE 在提取特征时考虑了整个质谱,并不像SVM-RFE 仅仅局限于几个谱峰,因此经过AP +SVM-RFE 处理得到的特征包含了更多有用信息,因而分类率也会更高。
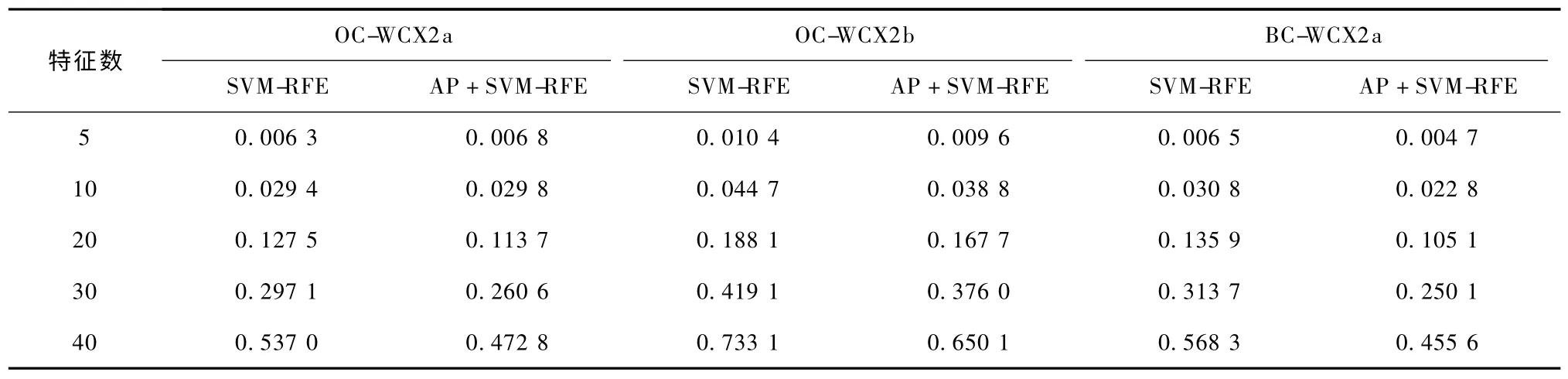
表1 3 个数据集中特征间的平均皮尔逊相关系数Tab.1 Average Pearson correlation coefficient between features in three datasets
2.3 挑选生物标志物
经过实验,AP +SVM-RFE 算法不仅从3 个数据集中得到分类率等结果,而且提取出特征作为生物标志物(biomarker,单位为Da),表2 为2 个数据集中提取出的10 个生物标识物。对于公共数据OCWCX2a 和OC-WCX2b,其中某些蛋白质特征位点已经发现,并用星号标记,其余为新发现的蛋白质特征位点。
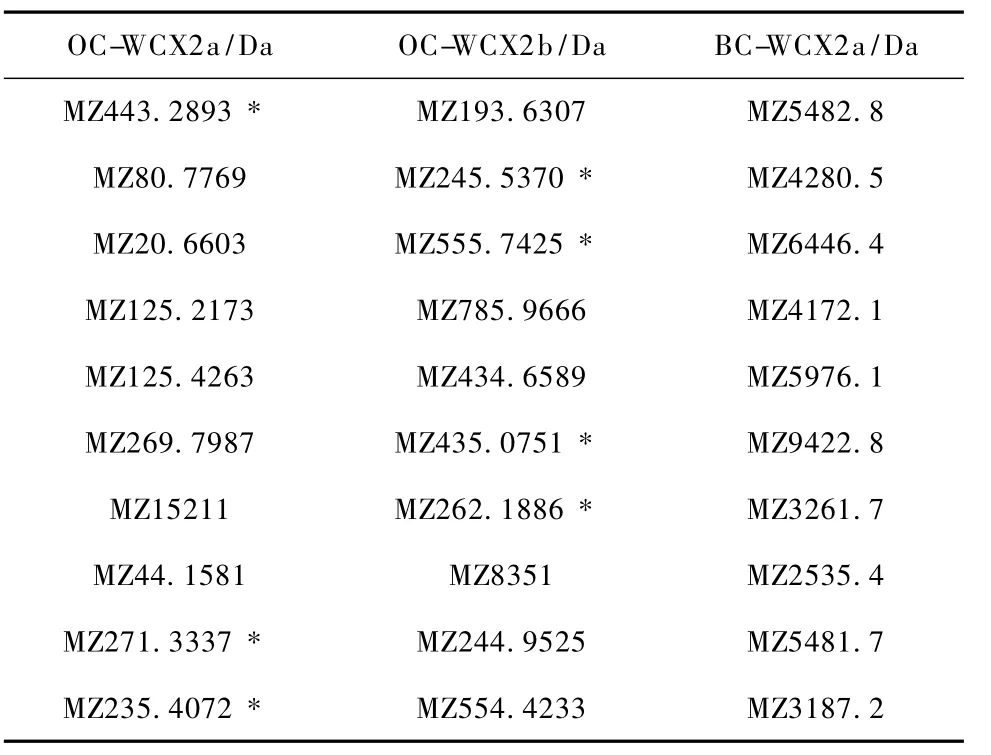
表2 算法在3 个数据集中提取的10 个特征蛋白位点Tab.2 10 differential m/z biomarkers for 3 datasets
3 讨论
由于基于蛋白质组学的质谱技术快速发展,使之成为癌症辅助诊断的有力工具。前人都提出了算法,且这些算法都取得了一定的实验效果,但是大部分关注的重点为算法对病例数据的分类效果,并没有筛选出与疾病相关的蛋白质位点。
本研究提出的算法不仅得到了分类效果,而且筛选出与疾病可能相关的蛋白质位点,对临床诊断具有一定的指导意义。分析SELDI-TOF 质谱数据的方法有很多,聚类分析是其中较为有效的一种方法。对于高通量SELDI-TOF 质谱数据以及基因微阵列数据,聚类分析算法在处理高通量数据方面已取得一定的进展。虽然有些聚类分析算法存在一些缺陷,但是在降低特征维数和去除特征间相关性方面,聚类分析不仅运算复杂性较低,而且取得了较好效果。
在实际临床诊断中,医学上通常更关注敏感性。由于将癌症患者误诊断为正常,会使病人丧失最佳治疗时期甚至导致死亡,而将正常人诊断为癌症患者可以在医学上进行进一步诊断,因此医学诊断更多强调敏感性。
此外,在医学诊断中关注的不仅仅是分类率、敏感性和特异性等值,最主要的是在高分类率条件下挑选出具有高判别效果的蛋白质位点。通过研究不同类型癌症所对应的蛋白质位点,即可对该类型癌症进行早期诊断,因此这些蛋白质位点对于医生诊断早期癌症患者具有一定的辅助作用。但是,由于现阶段基于蛋白质组学的质谱数据研究对于数据预处理参数的设定不尽相同,在临床中易受到外界条件的干扰,进而会影响到实验结果,所以单纯依靠质谱数据对于临床诊断并不完全可靠,提供的信息对于医学诊断来说也仅是参考,如果要完全判断患者情况还需要其他的手段,比如利用核磁共振成像(MRI)来诊断早期癌症。虽然现阶段基于蛋白质组学的质谱技术对于早期临床诊断癌症有一定困难,但随着研究的深入,以及近年来该领域取得的长足进步,相信在不远的将来基于蛋白质组学的质谱技术必定会成为癌症辅助诊断的强有力助手。
4 结论
本研究提出了一种针对SELDI-TOF 蛋白质质谱数据的特征选择算法。该算法以最终获得病例数据的分类效果和降低特征之间的相关性为目的,提取了3 个数据集的生物标志物。采用基于近邻传播聚类分析的蛋白质质谱数据特征选择算法,不但有效地降低了特征间的相关性,而且保留了最具有判别意义的特征。在公共卵巢癌数据集OC-WCX2a、OC-WCX2b 和浙江省肿瘤医院乳腺癌数据BCWCX2a 上的实验结果表明,该算法能够获得较高的分类率,并且发现新的蛋白质特征位点,这对临床的癌症诊断和发病机制的研究具有一定的意义。值得一提的是,本研究对蛋白质特征位点的选择仅仅依靠了信息学手段,至于挑选出的生物标志物在临床上是否有效,还有待于在临床上的检验。
[1] Jemal A,Sieg el R,Ward E,et al. Cancer statistics[J]. A Cancer Journal of Clinicians,2011,(61):212 -236.
[2] Bruce A,Julian L,Martin R,et al. Molecular biology of the cell,fourth edition[M]. New York:Garland Science,2004.
[3] Shin H,Sheu B,Joseph M,et al. Guilt-by-association feature selection:identifying biomakers from proteomic profiles [J].Biomed Inform,2008,41:124 -136.
[4] Somorjai RL,Dolenko B,Baumgartner R. Class prediction and discovery using gene microarray and proteomics mass spectroscopy data: curses, caveats, cautions [ J ].Bioinformatics,2003,19(12):1484 -1491.
[5] Thomas A,Tourassi GD,Elmaghraby AS,et al. Data mining in proteomic mass spectrometry[J]. Clinical Proteomics,2006,2(1):13 -21.
[6] Hanczar B,Courtine M,Benis A,et al. Improving classification of microarray data using prototype-based feature selection[J].SIGKDD Exploration,2003,5:23 -30.
[7] Wang Yuhang,Fillia Makedon,James Ford,et al. HykGene:a hybrid approach for selecting marker genes for phenotype classification using microarray gene expression data [J].Bioinformatics,2005,21(8):1530 -1537.
[8] Yang Pengyi,Zhang Zili,Zhou Bingbing,et al. A clustering based hybrid system for biomaker selection and sample classification of mass spectrometry data[J]. Neurocomputing,2010,73(13 -16):2317 -2331.
[9] Yang Pengyi,Zhang Zili. A clustering based hybrid system for mass spectrometry data analysis[J]. Pattern Recognition in Bioinformatics,Lecture Notes in Bioinformatics,2008,5265:98 -109.
[10] Huang Rui,Liu Qingshan,Lu Hanqing,et al. Solving the small sample size problem of LDA [J]. Pattern Recognition,2002,3:29 -32.
[11] Dueck D,Frey BJ. Clustering by passing messages between data points[J]. Science,2007,315(5814):972 -976.
[12] 李伟红,龚卫国,陈伟民,等. 基于SVMRFE 的人脸特征选择方法[J]. 光电工程,2006,33(5):113 -117.
[13] 王尧佳,祝磊,韩斌. 基于递归零空间线性判别分析算法的蛋白质质谱数据特征选择[J]. 航天医学与医学工程,2010,23(5):324 -328.
[14] 孟辉,洪文学. 蛋白质组学质谱数据预处理技术综述[J].中国生物医学工程学报,2009,28(3):469 -475.
猜你喜欢
食品安全导刊(2021年20期)2021-08-30
河南科学(2021年3期)2021-05-06
电子技术与软件工程(2019年18期)2019-11-18
质谱学报(2019年5期)2019-09-24
中国惯性技术学报(2018年4期)2018-11-08
电子技术与软件工程(2017年14期)2017-09-08
自动化学报(2017年5期)2017-05-14
真空与低温(2017年1期)2017-03-15
电子制作(2017年23期)2017-02-02
智能系统学报(2015年4期)2015-12-27
