
紫红笛鲷线粒体全序列与笛鲷属鱼类分化年代的贝叶斯分析
2013-03-07 07:12王中铎郭昱嵩刘楚吾
海洋科学 2013年1期
廖 杰, 王中铎, 郭昱嵩, 刘楚吾
(广东海洋大学 水产学院 南海水产经济动物增养殖广东普通高校重点实验室, 广东 湛江 524025)
紫红笛鲷线粒体全序列与笛鲷属鱼类分化年代的贝叶斯分析
廖 杰, 王中铎, 郭昱嵩, 刘楚吾
(广东海洋大学 水产学院 南海水产经济动物增养殖广东普通高校重点实验室, 广东 湛江 524025)
利用常规 PCR未纯化产物直接测序的方法, 获得紫红笛鲷(Lutjanus argentimaculatus)全长为16 543 bp线粒体DNA全序列, GenBank登录号为JN182927。结合GenBank中的已有数据比较分析表明: 紫红笛鲷与笛鲷属(Lutjanus)线粒体基因组成基本一致, 符合脊椎动物的特征, 各基因间进化速率差异显著; 13个蛋白编码基因拼接序列贝叶斯建树的后验率显著高于单基因或全序列, 更适用于笛鲷属的贝叶斯法系统分化分析和年代推断; 13个蛋白编码基因及其拼接序列进行的系统进化分析均支持紫红笛鲷与勒氏笛鲷(Lutjanus russellii)及海鸡母笛鲷(Lutjanus rivulatus)亲缘关系较近; 基于13个蛋白编码基因拼接序列的贝叶斯松散分子钟推算出笛鲷属鱼类两个主要支系大约在 61.5Ma前古新世的达宁阶与蒙蒂阶交界时期发生分化。
紫红笛鲷(Lutjanus argentimaculatus); 线粒体全序列; 贝叶斯分析; 松散分子钟; 笛鲷属鱼类
紫红笛鲷(Lutjanus argentimaculatus)隶属硬骨鱼纲(Osteichthyes), 鲈形目(Perciformes), 笛鲷科(Lutjanidae), 笛鲷属(Lutjanus)。主要分布于印度洋和太平洋中部及西部, 包括中国的南海和东海南部, 是一种优质的食用鱼类。有关紫红笛鲷的研究很多, 涉及人工养殖、种群遗传多样性以及它在笛鲷属中的关系等多个方面。邢玉娜等[1]、区又君等[2]分别研究了紫红笛鲷育苗和幼鱼养殖密度与摄食关系; 张俊彬等[3]研究了中国南海部分地区紫红笛鲷养殖和野生群体的遗传多样性。Russell等[4]综述了澳大利亚紫红笛鲷生态特征、群体遗传结构以及养殖技术, 并记录紫红笛鲷存在独特的生活史:幼鱼和早期成鱼都可以生活在淡水溪流中。Guo等[5]基于线粒体基因(COX2和Cytb)的系统进化表明紫红笛鲷与红鳍笛鲷(Lutjanus erythopterus)关系密切;王中铎等[6]综合利用3个线粒体基因(COX1、COX2和Cytb)和2个核基因(RAG1和RAG2)构建了笛鲷属鱼类的系统进化树, 揭示出紫红笛鲷在进化上最早与大体型、红体色、近岸底栖的红鳍笛鲷和千年笛鲷(Lutjanus sebae)一支分化, 独立于其他笛鲷属鱼类, 自成一支。
近年来发展起来的贝叶斯模型参数可直接进行量化,并以后验概率来表示各分支的可信度而不需用自引导法(Bootstrap)进行检验, 而且在计算速度和可靠性上明显优于其他算法, 十分利于线粒体全序列等较大数据集的分析[7-8]。 另外, 贝叶斯松散分子钟利用化石标定点计算分化时间, 推测出实验对象出现或发生年代已经引起鱼类进化研究者的广泛重视[9-11]。随着测序技术的进步使线粒体全序列的获得变得越来越容易, 用线粒体全序列作为遗传进化分析的材料, 将会获得更加全面的信息进而得到更合理的结论, 这一技术在鱼类进化研究中的应用越来越广泛[12-14]。一直以来对笛鲷属鱼类的分类学地位和形态分类学标准都存在争议[6,11,13]。Guo等[5-6]利用线粒体基因(COX2和Cytb)和核基因重建了笛鲷属的系统进化历程, 并且先后获取了多种笛鲷属鱼类的线粒体DNA(mtDNA)全序列。
本研究通过测定紫红笛鲷全序列进而确定该物种在笛鲷属的分类地位, 并通过贝叶斯分析推断笛鲷属各支系的分化年代同时分析比较线粒体全序列及其各编码区段贝叶斯建树的优劣性, 为揭示鱼类演化研究积累基础资料。
1 材料与方法
1.1 实验材料和总DNA的提取
紫红笛鲷0.8 kg, 性腺发育未成熟, 购自湛江市霞山区水产品交易市场。采用形态与DNA 条码结合的方法鉴定实验对象[14], 参照卢圣栋等[15]的方法从肌肉或鳍条中提取样本DNA, 4℃保存备用。
1.2 LA-PCR纯化线粒体DNA
引物参考王中铎等[10](表 1), 均由上海 Sangon生物技术公司合成。采用长距 PCR(LA-PC R)引物LA1和LA2扩增得到覆盖整个线粒体DNA序列的大片段, PCR反应体系为: 总体积25 μL, 含1×PCR LA Buffer, dNTPs 15 nmol, 引物各 20 pmol, LATaqDNA聚合酶(TaKaRa) 2.5U, 总DNA 约20 ng。扩增条件: 94 ℃预变性1 min; 98℃变性10 s, 68℃退火16 min, 循环30次; 最后72℃延伸10 min。1%的琼脂糖电泳对扩增产物进行检测。
1.3 常规PCR扩增获取线粒体DNA片段
以稀释20倍的LA-PCR 产物为模板, 使用表1所列常规PCR引物扩增获取长度600~1 000 bp左右的相互重叠的小片段。反应体系为: 总体积50 μL, 含1×PCR Buffer, dNTPs 10 nmol, 引物各20 pmol, rTaqDNA聚合酶(TaKaRa) 2.5 U, DNA约20 ng。PCR 反应的循环参数为: 94 ℃预变性3 min, 94 ℃变性42 s, 50~55 ℃退火30 s, 72 ℃延伸55 s, 共30个循环, 最终72 ℃延伸10 min, 用1.0%琼脂糖凝胶电泳检测扩增产物, 凝胶成像仪拍照。PCR扩增产物委托广州华大基因公司进行序列双向测定。
1.4 全序列拼接与分析
用Invitrogen软件包的ContigExpress进行序列拼接, 利用Editseq程序(http://www.dnastar. com/)查找序列进行开放阅读框, 获得序列互补链以及统计各密码子所占比例; 利用 MEGA 4.0 软件完成蛋白编码序列的翻译和氨基酸编码情况的调查[16]; 在线查找tRNA (http://lowelab.ucsc.edu/tRNAscan-SE)[17];用 Sequin 软件(http://www.ncbi.nlm.nih.gov/Sequin/ index.html)递交序列至 GenBank。从 GenBank下载的其余7种笛鲷的线粒体全序列(6种为本研究室测定)。分别利用ATPase6、Cytb、COX1、COX2、ND1、ND2、ND4、ND5等基因以及D-loop区进行遗传距离分析。
1.5 基于线粒体全序列和编码基因及其拼接序列的贝叶斯分析
参考Drummond等[18]的方法利用Beast 1.6.1软件包(http://beast.bio.ed.ac.uk/)构建贝叶斯树, 并利用贝叶斯松散分子钟(uncorrected relaxed lognormal clock)推断分化年代[19]。软件运行设置如下: (GTR+G+I+relaxed, MCMC=1000000, burn in=1000)利用 FigTree v1.3.1(http://tree.bio.ed.ac.uk/)生成树图。所选的外群物种为斑马鱼(Danio rerio)和青鳉(Oryzias latipes), 化 石 标 定 点 则 为 鲀 科(Tetraodontidae)鱼类红鳍东方豚(Takifugu rubripes)和凹鼻鲀(Tetraodon nigroviridis), 它们的分化上下限为56.0 ~32.25 Ma[20]。
1.6 不同基因贝叶斯后验概率分析
分别建立全序列、13个蛋白编码基因及其拼接序列的贝叶斯分析树, 以笛鲷主分支, 紫红笛鲷所在支以及所有分支平均贝叶斯后验概率最为判断各基因和拼接序列贝叶斯建树的优劣性。
2 实验结果
2.1 片段扩增、序列拼接及分析
LA-PCR获得的长距产物(图1)为模板进行常规PCR(图2), 扩增产物直接测序后拼接获得16 543 bp的紫红笛鲷线粒体全序列。序列中(A、T、C、G)4种碱基分别占 28.25%, 24.68%, 30.95%, 16.12%, A+T>G+C。用Sequin上传至Gene Bank, 登录号为JN182927。紫红笛鲷mtDNA基因组包括13个蛋白编码基因, 22个tRNA基因, 2个rRNA基因和1个控制区。基因分布呈不均一性, 其中轻链(L-strand)仅编码8个tRNA基因(Pro、Glu、Ser、Tyr、Cys、Asn、Ala、Gln)及ND6基因, 其余基因皆由重链(H-strand)编码。
2.2 与已有笛鲷线粒体全序列的比较分析
分析得到紫红笛鲷与7种笛鲷的ATPase6、Cyt b、COX 1、COX 2、ND 1、ND 2、ND 4、ND 5、D-loop和线粒体全序列的遗传距离(表 2)。从表中可见, 单基因的物种间遗传距离平均值为线粒体全序列间的1.4~1.9倍。基于基因片段所得的K2P遗传距离平均值比较可以看出: 紫红笛鲷与马拉巴笛鲷(Lutjanus malabarius)、红鳍笛鲷和千年笛鲷的距离较远(≥0.239), 而与其他笛鲷相对较近(≤0.195); 其中在

表1 线粒体DNA基因组序列测定引物Tab. 1 PCR primers for mtDNA complete genome
ATPase6间的遗传距离比较中紫红笛鲷与其他 4种笛鲷的遗传距离值比其他基因片段的大, 而线粒体全序列间遗传距离比较的结果与 12个基因片段的相似,均为马拉巴笛鲷、红鳍笛鲷和千年笛鲷的遗传距离较远。所有基因中海鸡母笛鲷(L. rivulatus)的距离都最小,它相对其他笛鲷与紫红笛鲷的关系更为密切。
图1 LA-PCR电泳检测图Fig. 1 Electrophoresis of LA-PCR
图2 部分常规PCR电泳检测图Fig. 2 Electrophoresis of common PCR
2.3 基于贝叶斯松散分子钟的笛鲷分化年代推断分析
分别建立 8种笛鲷线粒体全序列、线粒体各个编码基因以及线粒体COX1基因的贝叶斯分析树,得到多个贝叶斯推断图(图3~图5)。3图中标定化石鲀科(Tetraodontidae)种都落在化石记录范围的中心。基于线粒体全序列的推断结果显示笛鲷属的分化年代处于40.86Ma, 而基于13个蛋白编码基因拼接序列的分析则显示分化发生在 61.49Ma, 两者推断的主分支年代差异很大。图 3中个小分支发生都位于32.7~8.2Ma这个相对较短的时间, 而图 4中各小分支的出现年代跨度则小得多(57.8~48.7Ma)。两个系统进化树中8种笛鲷都可以明确的划分成两个支系。进化树和遗传距离各自的比较结果相吻合。遗传距离较远的红鳍笛鲷, 马拉巴笛鲷和千年笛鲷聚为一支, 紫红笛鲷所在一支的另五种笛鲷聚为另一支。
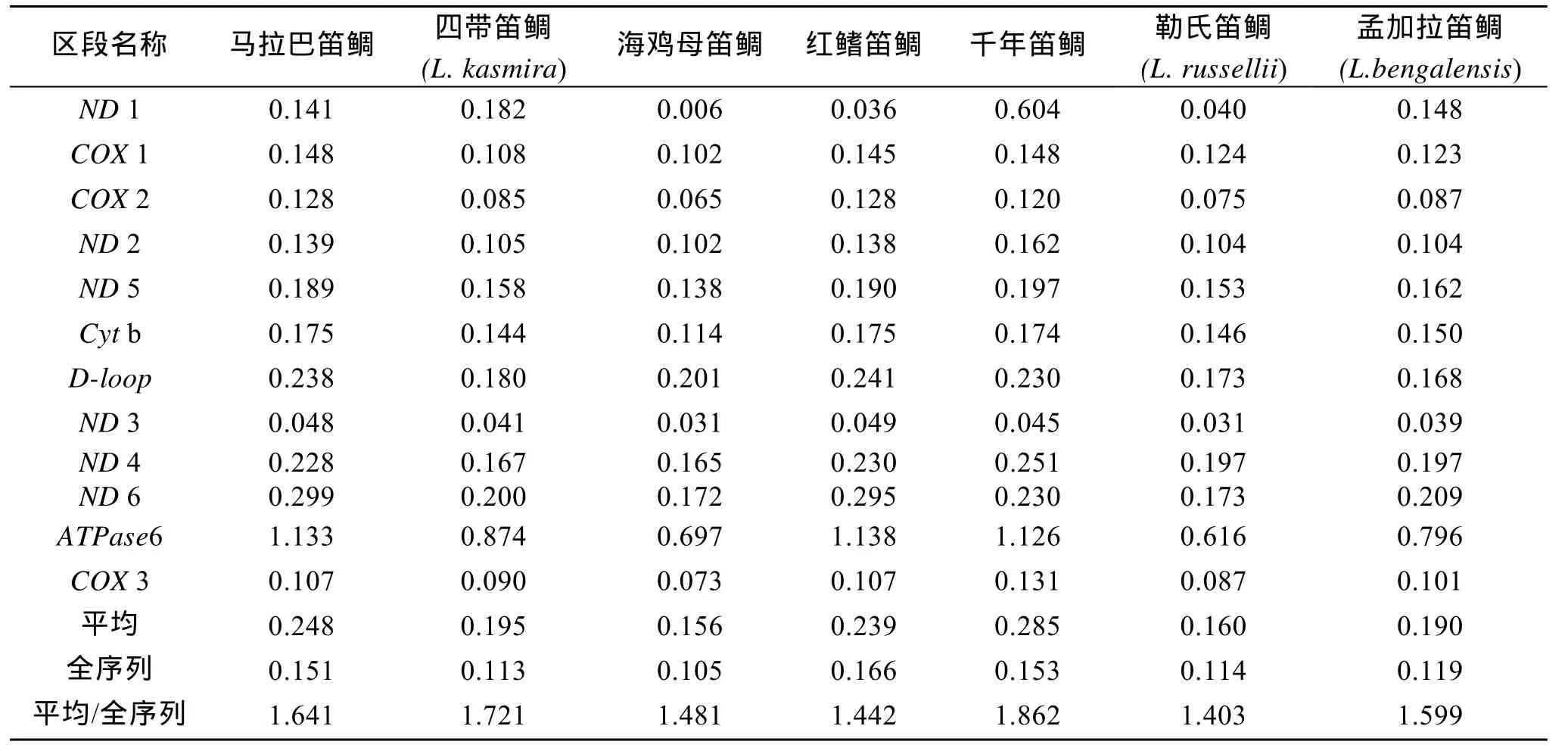
表2 K2P紫红笛鲷与7种笛鲷各基因区段遗传距离比较Tab. 2 K2P Genetic distances between Lutjanus argentimaculatus and other snappers
在 3个图中紫红笛鲷所处的位置存在差异, 基于线粒体全序列的贝叶斯进化树笛鲷分支不够细致,而另外两个图中各个小支明显分开。图3和图5中紫红笛鲷和海鸡母笛鲷(Lutjanus rivulatus)关系较紧密, 图4中紫红笛鲷和勒氏笛鲷(Lutjanus russellii)先聚为一支再和海鸡母笛鲷并列。
线粒体DNA全序列、13个基因拼接序列以及单独的基因序列建立贝叶斯分析树, 笛鲷主分支、紫红笛鲷分支以及平均后验概率统计如表 3所示。基于mtDNA蛋白编码基因拼接序列的贝叶斯分析, 无论是各支还是总后验概率上都是最高点, 而基于线粒体全序列贝叶斯分析树的各支后验概率都很低。不同 mtDNA蛋白编码基因得出的后验概率差别很大,COX1、ND2、ATPase6总后验概率最高,ND3、ND4、ND4L、COX3中等,ND1、ND5、ND6、COX2、Cytb、ATPase8较差,COX1、ND2、ATPase63个基因的拼接序列后验概率也相对较高。紫红笛鲷所在支的后验概率在0.9以上的有ND2、ND3、ND4L树图均显示紫红笛鲷和勒氏笛鲷聚为一小支, 这和13个mtDNA蛋白编码基因拼接序列聚类一致。
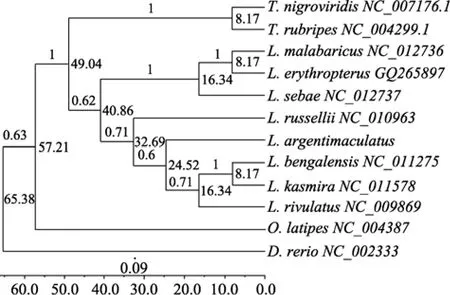
图3 笛鲷线粒体全序列贝叶斯树图Fig. 3 Bayesian evolutionary tree of Mitochondrial complete sequence
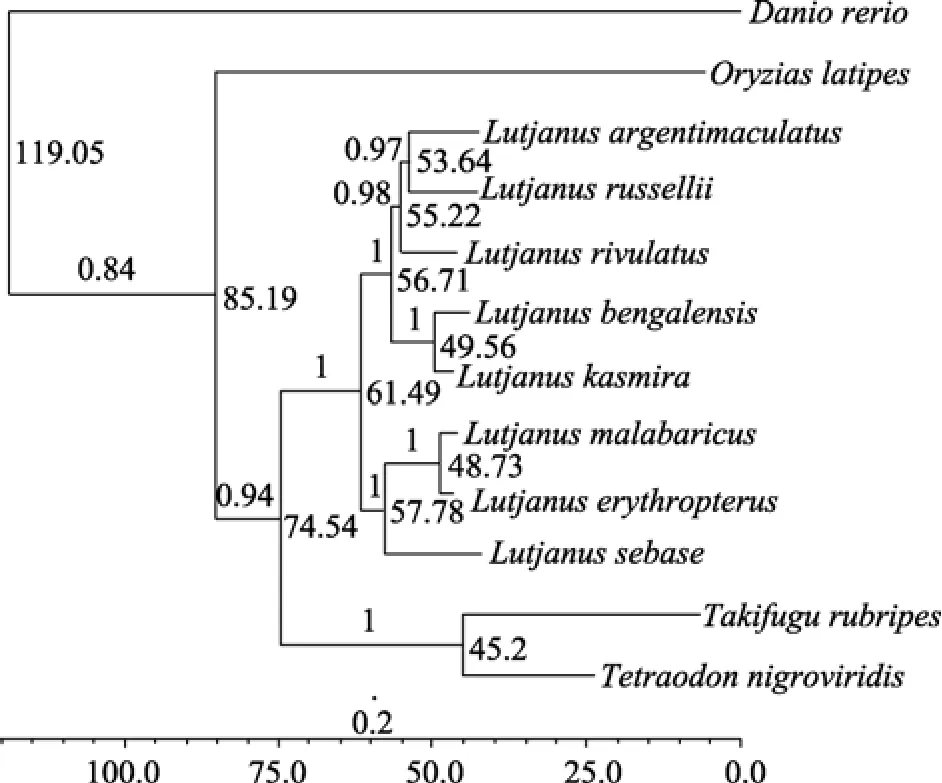
图4 线粒体蛋白编码序列基因贝叶斯树Fig. 4 Bayesian evolutionary tree of Mitochondrial protein coding gene
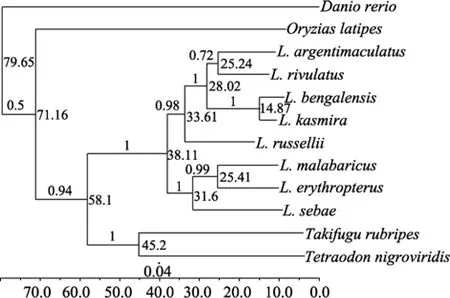
图5 笛鲷属鱼类COX 1基因全序列贝叶斯树Fig. 5 Bayesian evolutionary tree of COX 1 complete sequences of snappers
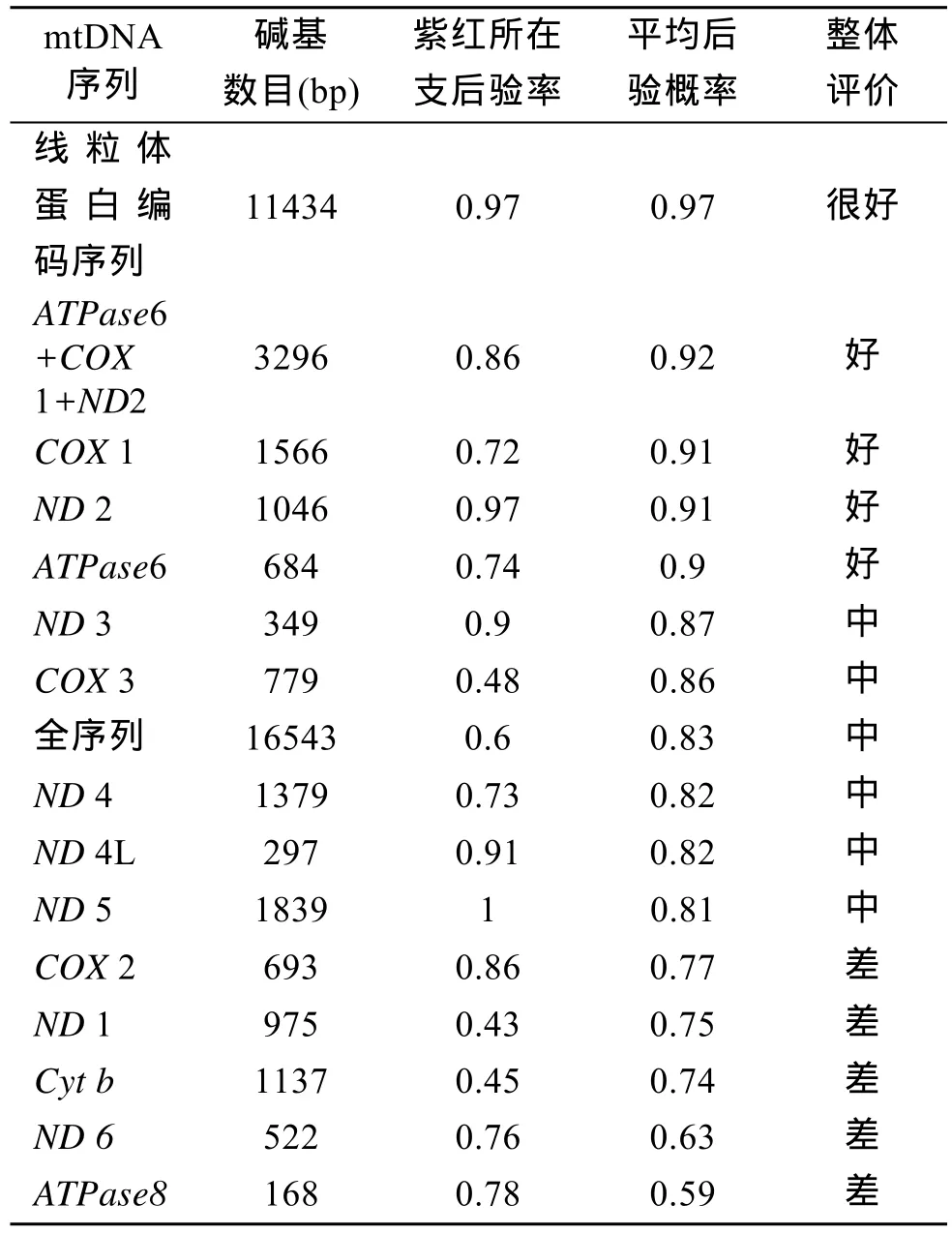
表3 线粒体DNA部分区段贝叶斯后验概率Tab. 3 Bayesian posterior probability of mtDNA partial genes region
3 分析与讨论
本研究基于获取的紫红笛鲷 mtDNA全序列和已存在的笛鲷mtDNA全序列, 探讨了适用于贝叶斯系统进化分析的最佳序列, 明确了紫红笛鲷与其他7种笛鲷的进化关系, 并推断了笛鲷两大支系的分化年代。
3.1 线粒体DNA全序列的高效获取与分析
相比以直接提取获得的总DNA为模板, 运用长距PCR的方法获得紫红笛鲷线粒体DNA长距片段,保证了总DNA很少量时模板量足够使用, 同时提高了模板质量, 采用常规 PCR产物直接送测, 测序结果分析简便, 整个周期短[10,12]。此方法可方便、快速获得高质量的线粒体全序列。之前对笛鲷鱼类研究一般常用 MEGA进行建树分析[6,24], 这种方式只能获得物种间的进化关系, 不能获得分化年代方面的结果。本研究尝试运用BEAST 软件对笛鲷全序列、mtDNA编码区基因、mtDNA编码区基因拼接数据集进行了贝叶斯分析, 并采用松散分子钟模型推断分歧的发生时间[21], 与MrBayes相比[22], BEAST操作直观, 而且配套有 Figtree程序, 数据分析参数可进行及时调整, 生成的进化树和进化年代可进行方便地编辑和标记。
3.2 基于贝叶斯法的最佳 mtDNA区段选择
对进化关系研究时线粒体基因组 DNA及其 13个编码基因的适用性, 以往研究者已经进行了一系列基于NJ等传统方法的分析。Zardoya等[23]比较了不同基因区分脊椎动物门下的各纲的能力, 认为COX1、Cytb、ND2、ND4、ND5都非常适合纲之间进化研究; 陈姝君等[24]比较了硬骨鱼类目间不同基因的适应性, 认为COX1、COX3、ND4、ND5最为出色; 郭昱嵩[25]则认为COX 2、ND 6最为适合鲈形目各属间进化分析。王中铎[6]对笛鲷属的研究揭示出Cytb、COX1、COX2是最适合系统进化分析的单基因, 13个蛋白编码基因拼接得到组合片段建立的NJ树的置信度比单个基因有显著提高, 但低于线粒体全序列。可见, 基于分类阶元不同, 区段的适用性存在差异。
本研究运用贝叶斯法分析了笛鲷属系统发生,平均后验概率的高低次序为: 13个蛋白编码基因拼接序列(0.97)>COX1+ND2+ATPase6(0.92)>COX1、ND2、ATPase6、ND3和COX3(0.91~0.86)>全序列(0.83)>余下 8个单基因(0.82~0.59)。这与王中铎等NJ法进行系统进化分析时的最佳线粒体区段存在差异, 同时他的研究的结果表明全序列会优于任一编码区。全序列的贝叶斯后验率明显低于13个蛋白编码基因拼接序列, 甚至低于COX1、ND2、ATPase6等基因, 以 13个蛋白编码基因拼接序列进行贝叶斯法年代推断比全序列更可信。
3.3 笛鲷属分化年代的贝叶斯法推断
本研究通过紫红笛鲷和各种笛鲷遗传距离比较分析及贝叶斯进化树分析表明, 虽然紫红笛鲷在形态上属于大型笛鲷种, 但是其生活史表现出特殊性[4]。表2中K2P遗传距离统计显示紫红笛鲷和另外3种大型笛鲷(马拉巴笛鲷、千年笛鲷、红鳍笛鲷)遗传距离较远, 而与勒氏笛鲷、海鸡母笛鲷较近。贝叶斯树图更直观地表明这种关系。
Bruno等[26]研究发现贝叶斯法推断系统发育往往会高估节点的后验概率, 认为只有后验概率大于95%的节点才是可信的。基于全序列和不同基因的分析中各支的后验概率都未达到这一标准, 基于 13个蛋白编码基因拼接的贝叶斯分析中各个节点的后验率均大于95%, 支持紫红笛鲷与勒氏笛鲷聚类。基于mtDNA的13个编码区拼接序列, 贝叶斯松散分子钟推测笛鲷属两大类群的分化年代距今约在61.5Ma年左右, 根据世界地质年代表记录这一时期处于古新世期间(65~53 Ma)的达宁阶与蒙蒂阶交界时期。
[1] 邢玉娜, 尹绍武, 陈国华,等. 紫红笛鲷Lutjanus argentimaculatus(Forsska)繁殖和池塘育苗研究[J]. 现代渔业信息, 2005, 8: 25-27.
[2] 区又君, 勾效伟, 廖锐, 等. 紫红笛鲷幼鱼对集群密度和投饵定额的摄食反应[J]. 动物学杂志, 2008, 43(3): 34-38.
[3] 张俊彬, 黄良民, 陈真然. 紫红笛鲷遗传多样性的AFLP分析[J]. 海洋学报. 2005, 3, 27(2): 165-171.
[4] Russell D J, McDougall A J, Fletcher A S, et al. Biology, management and genetic stock structure of mangrove jack (Lutjanus argentimaculatus) in Australia[J]. FRDC, 2003, 18(9): 1456-1563.
[5] Guo Y S, Wang Z D, Liu C W, et al. Sequencing and analysis of the complete mitochondrial DNA of Russell’s snapper (L. russellii) [J]. Natural Science, 2008, 18: 1233-1238.
[6] 王中铎, 谭围, 郭昱嵩, 等.笛鲷属线粒体基因组编码序列的系统进化分析能力评估[J]. 水产学报, 2010, 34(6): 656-663.
[7] 张艳春. 大口鳒Psettodes erumei线粒体全序列的研究和鲽形目鱼类系统进化分析[D]. 青岛: 中国海洋大学, 2009.
[8] 郑楠, 张原, 敖滨, 等. 贝叶斯推论重建乳头瘤病毒的进化系统[J]. 病毒学报, 2002, 19(4): 293-300.
[9] Kawahara R, Miya M, Mabuchi K, et al. Interrelationships of the 11 gasterosteiform families (sticklebacks, pipefishes, and their relatives): A new perspective based on whole mitogenome sequences from 75 higher teleosts [J]. Molecular Phylogenetics and Evolution, 2008, 46: 224-236.
[10] 王中铎, 郭昱嵩, 刘楚吾, 等. 军曹鱼线粒体 DNA全序列与 鲹鱼宗系的系统进化[J]. 水生物学报, 2011, 35(2): 1-9.
[11] 李志强, 郭宝成, 李俊兵, 等. 贝叶斯联合模型与中国洞穴鱼类分化时间的估算[J]. 科学通报, 2008, 53(13): 1560-1569.
[12] 谭围, 王中铎, 郭昱嵩, 等. 孟加拉笛鲷线粒体基因组序列结构及其进化[J]. 中国生物化学与分子生物学报, 2009, 25(3): 287-291.
[13] Miya M, Nishida M. Use of mitogenomic information in teleostean molecular phylogentics: a tree-based exploration under the maximum-parsimony optimality criterion[J]. Molecula Phylogenetics and Evolution, 2000, 17(3): 437-455.
[14] 王中铎, 郭昱嵩, 刘楚吾, 等. 笛鲷属鱼类DNA分子条码、系统进化和成种机制[J]. 中国科学, 2010, 6: 516-521.
[15] 卢圣栋. 现代分子生物学实验技术[M]. 北京: 中国协和医科大学出版社, 1999: 6.
[16] Kumar S, Nei M, Dudley J, et al. MEGA: a biologist-centric software for evolutionary analysis of DNA and protein sequences[J]. Brief Bioinform, 2008, 9(4): 299-306.
[17] Lowe T M, Eddy S R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence[J]. Nucleic Acids Research, 1997, 25(5): 955-964.
[18] Drummond A J, Rambaut A. BEAST: Bayesian evolutionary analysis by sampling trees[J]. BMC Evolutionary Biology, 2007, 7: 214.
[19] Drummond A J, Ho S Y, Phillips M J, et al. Relaxed phylogenetics and dating with confidence[J]. PLoS Biology, 2006, 214(7):699-710.
[20] Benton M J, Donoghue P C J. Paleontological evidence to date the tree of life [J]. Molecular Biology and Evolution, 2007, 24: 26-53.
[21] Leache A D, Reeder T W. Molecular systematics of the Eastern Fence Lizard (Sceloporus undulatus): a comparison of parsimony, likelihood, and bayesian approaches[J]. Systematic Biology, 2002, 51: 44-68.
[22] Ronquist F, Huelsenbeck J P. MrBayes 3: Bayesian phylogenetic inference under mixed models[J]. Bioinformatics, 2003, 19: 1572-1574.
[23] Zardoya R, Meyer A. Phylogenetic performance of mitochondrial protein-coding genes in resolving genes in resolving relationgships among vertebrates[J]. Molecular Phylogenetic and Evolution, 2000, 17(3): 437-455.
[24] 陈姝君, 赫崇波, 木云雷, 等.硬骨鱼类线粒体基因系统发育信息效率分析[J].中国水产科学, 2008, 15(1): 12-21.
[25] 郭昱嵩, 王中铎, 焦阳, 等. 鲈形目线粒体DNA蛋白编码基因的适应性分析[J]. 自然科学进展, 2008, 18(9): 7355-7358.
[26] Bruno W J. Weighted neighbor joining, a likelihood-based approach to distancebased phylogeny reconstruction[J]. Mol Biol, 2000, 17(4): 189-197.
(本文编辑: 谭雪静)
Complete mitochondria sequence ofLutjanus argentimaculatusand Bayesian analysis
LIAO Jie, WANG Zhong-duo, GUO Yu-song, LIU Chu-wu
(Fisheries College, Key Laboratory of Aquaculture in South China Sea for Aquatic Economic Animal of Guangdong Higher Education Institutes, Guangdong Ocean University, Zhanjiang 524025, China)
Dec.,30,2011
Lutjanus argentimaculatus; mitochondrion genome; bayesian analysis; uncorrected relaxed lognormal clock; snapper fish
We got 16 543 bp Red snapper (Lutjanus argentimaculatus) complete mitochondrial DNA sequence through common PCR, with the GenBank registration number being JN182927. Our sequence and the data in Gen-Bank, red snapper’s mitochondrion gene composition were consistent with other snappers, according to the characteristics of vertebrates. Bayesian tree of complete sequence and 13 protein-coding genes and their splicing sequence indicated that: red snapper was close toL. russelliiandL. rivulatus. The evolution rate differences of different mitochondrial sections were obvious.COX1,ND2 andATPase6 Bayesian posterior rates were significantly higher than the other ten in 13 protein-coding mitochondrial genes of. The 13 protein-coding gene splicing sequences Bayesian posterior rates were significantly higher than completed sequence, and were more suitable for Bayesian uncorrected relaxed lognormal clock analysis. Thirteen protein coding gene splicing sequence’s Bayesian uncorrected relaxed lognormal clock work out snappers’ two main branches broke out in 61.5 Ma before, which was the junction point of Danian and Selandian in Paleocene.
Q349
A
1000-3096(2013)01-0062-08
2011-12-30;
2012-04-06
国家自然科学基金项目(31101904, 31201996); 广东省自然科学基金项目(10452408801004224)
廖杰(1986-), 男, 湖南湘潭人, 硕士, 主要从事海洋生物研究工作, 电话: 13809887920, E-mail: liaojie05102@163.com;王中铎,
, 电话: 13659795480, E-mail: aduofa@gmail.com
猜你喜欢
昆明医科大学学报(2021年8期)2021-08-13
工程数学学报(2020年3期)2020-07-06
长治学院学报(2019年2期)2019-07-24
雷达学报(2017年6期)2017-03-26
文学少年(有声彩绘)(2017年7期)2017-02-17
铁道通信信号(2016年6期)2016-06-01
北京信息科技大学学报(自然科学版)(2016年6期)2016-02-27
电子器件(2015年5期)2015-12-29
天然产物研究与开发(2014年5期)2014-04-27
郑州大学学报(理学版)(2014年2期)2014-03-01
