
HPLC法测定蒙药嘎古拉-11中盐酸麻黄碱含量
2013-01-31 08:40杨光
中国民族医药杂志 2013年4期
杨 光
(内蒙古赤峰市医院,内蒙古 赤峰 024000)
HPLC法测定蒙药嘎古拉-11中盐酸麻黄碱含量
杨 光
(内蒙古赤峰市医院,内蒙古 赤峰 024000)
目的:测定蒙药嘎古拉-11中盐酸麻黄碱含量。方法 :HPLC法,流动相:乙腈-0.1%磷酸(5:95),波长:207nm,室温测定。结果:盐酸麻黄碱在0.0214μg~0.214μg范围内呈良好的线性关系。回归方程为Y=2258708.3X-673.2,r=1.000,线性关系良好。 3批次平均含量分别为:0.373、0.407、0.382(mg·g-1)。结论:本法方法测定,简便、易行,结果可靠。
嘎古拉-11;盐酸麻黄碱;HPLC法;含量测定
嘎古拉 -11由草果仁、紫草茸、木鳖仁(制)等 11味药组成,具有清热、消食功能。其中麻黄为方中君药,功能温中散寒,下气止痛。主要有效成分为1-麻黄碱,d-伪麻黄碱等。参照《中国药典》2005年版一部“麻黄”含量测定项下麻黄的测定方法,采用HPLC法对本品中的麻黄建立含量测定方法,通过实验摸索,确定了色谱条件,并经过方法学考察表明该方法操作简单,重现性好,专属性强。
1 仪器与试药
1.1 仪器:LC10-ATVP型高效液相议,CLASS-VP色谱工作站。岛津UV-2450型紫外-可见分光光度仪。
1.2 试剂与试药:对照品 盐酸麻黄碱(C10H15NO·HCL)(批号:171241-200303)中国药品生物制品检定所提供;供试品 由内蒙古赤峰市翁牛特旗中蒙医院提供。乙腈为色谱纯,水为超纯水,其它试剂为分析纯。
2 方法学考察
2.1 色谱条件的选择
2.1.1 色谱柱:色谱柱填充剂为十八烷基硅烷键合硅胶,本实验采用迪玛公司Diamonsil(钻石)色谱柱 C18(250×4.6mm,5μm)。
2.1.2 流动相的选择:参照《中国药典》2010年版一部“麻黄”含量测定项下麻黄的测定方法,用乙腈-0.1%磷酸为流动相,通过不同比例的比较,当流动相比例为(5:95)时,样品中的盐酸麻黄碱与其它成分达到较好的分离,并具有适宜的保留时间而且理论板数高,故确定流动相为乙腈 -0.1%磷酸(5:95)。
2.1.3 柱温:由于在室温条件下盐酸麻黄碱峰的保留时间稳定,且分离效果较好,故选定柱温为室温。
2.1.4 检测波长的选择:精密称取盐酸麻黄碱对照品适量,用甲醇制成 20μg·mL-1的溶液,在 200nm~600nm波长范围内扫描,结果盐酸麻黄碱在207nm处有最大吸收,结合《中国药典》2010年版一部“麻黄”含量测定项下盐酸麻黄碱的测定方法,选用207nm作为检测波长。
2.1.5 理论板数的确定:从对多批检测数据可见,盐酸麻黄碱的理论板数在3000以上,即能达到较好的分离效果,故确定理论塔板数按盐酸麻黄碱峰计算应不低于3000。
2.2 提取溶剂及提取时间的选择
2.2.1 提取溶剂的选择:参照《中国药典》2010年版一部“麻黄”含量测定项下盐酸麻黄碱的测定方法,选用甲醇作为提取溶剂。
2.2.2 提取效率的考察:以甲醇作为提取溶剂进行超声提取(功率 200W,频率 50kHz),为保证被测成分提取完全,实验中考察了模拟样超声提取 25、35、45、55(min)不同超声提取时间对提取效率的影响,实验表明超声处理45 min、55 min盐酸麻黄碱含量基本一致,故超声提取时间定为45 min。
2.2.3 中性氧化铝用量及洗脱液收集量的考察:精密量取上述提取后样品续滤液1mL,置中性氧化铝柱(100~200目,内径 1cm)中,用 50%甲醇洗脱,收集洗脱液置10mL量瓶中,为保证被测成分洗脱完全,实验中考察了氧化铝用量及洗脱液收集量对提取效率的影响,结果表明:中性氧化铝用量1.5g以下含量基本稳定不变,故将中性氧化铝用量定为1.5g。洗脱液收集量在9mL以上含量基本稳定不变,故将洗脱液收集量定为9mL。
2.3 溶液的制备:对照品溶液的制备:精密称取盐酸麻黄碱对照品适量,加甲醇制成每1mL含0.1mg的溶液,精密量取上述溶液2.0mL,置25mL量瓶中,用甲醇稀释至刻度,即得。
供试品溶液的制备:取本品适量,研细,取细粉2.5g,精密称定,置具塞锥形瓶中,精密加入甲醇 50mL,称定重量,超声处理(功率 200W,频率 50kHz)45分钟,放冷,再称定重量,用甲醇补足减失的重量,摇匀,滤过,精密量取 1mL,置中性氧化铝柱(100~200目,1.5g,内径 1cm)上,用 50%甲醇洗脱,收集洗脱液约 9mL,置10mL量瓶中,加磷酸1滴,用 50%甲醇稀释至刻度,摇匀,即得。
阴性对照溶液的制备:按处方比例并以相同工艺制备的缺麻黄的阴性对照,按供试品溶液制备法制得阴性对照溶液。
按上述色谱条件,分别精密吸取对照品溶液、阴性对照溶液、供试品溶液各 10μL测定,结果阴性对照色谱图在与盐酸麻黄碱对照品以及供试品色谱图相应的保留时间处无色谱峰出现,表明成方制剂中其它组分对测定无干扰。
2.4 线性关系考察:取盐酸麻黄碱对照品(批号:171241-200303)约 10mg,置 100mL量瓶中,加甲醇使溶解并稀释至刻度,摇匀,精密吸取 0.5mL、1.0mL、2.0mL、4.0mL、5.0mL,分别置 25mL量瓶中,加甲醇稀释至刻度,摇匀,各取10μ l注入液相色谱仪,按上述色谱条件测定,以峰面积对进样量进行回归分析,结果盐酸麻黄碱在0.0214μg~0.214μg范围内呈良好的线性关系。回归方程为 Y=2258708.3X-673.2,r=1.000,线性关系良好。
2.5 样品溶液稳定性试验:取同一份供试品溶液,分别于 0h、2h、4h、16h、24h进行了测定,记录峰面积,求得盐酸麻黄碱的RSD为 0.29%,24h内峰高无明显变化,表明供试品溶液制备24h内是基本稳定的。
2.6 精密度:重复性试验:取同一批号供试品 6份,各约2.5g,精密称定,分别按含量测定项下方法操作,测定每份供试品含量,测得盐酸麻黄碱含量RSD为0.18%。
2.7 准确度:加样回收试验:精密称取盐酸麻黄碱对照品 10.5mg(批号:171241-200303),置 100mL量瓶中,加甲醇使溶解并稀释至刻度,摇匀,备用。取同一批号的供试品9份,精密称定,分别置9个锥形瓶中,其中 3个锥形瓶中精密加入上述对照品溶液7mL;另3个锥形瓶中精密加入上述对照品溶液10mL;其余3个锥形瓶中精密加入上述对照品溶液12mL。按上述方法,精密加甲醇使总体积为25mL,同法操作,测定每份供试品的含量,计算平均回收率为99.6%,RSD为1.84%。
2.8 样品含量测定:取对照品及供试品溶液各10μL测定,结果见表1。
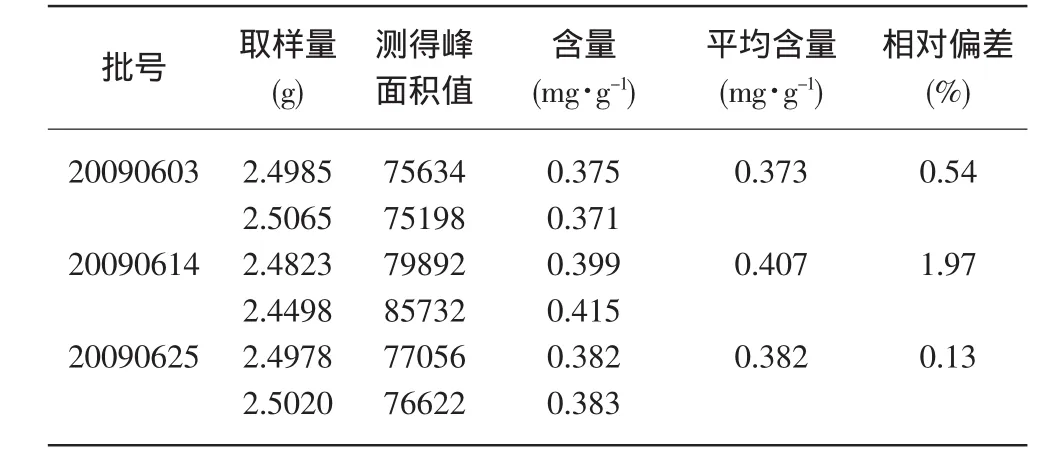
表1 样品中盐酸麻黄碱含量测定结果
3 讨论
通过以上实验可见,采用此方法测定,简便、易行,结果可靠。提取工艺简单,使被测成分在提取过程中出现的偶然误差减少,所得结果也相对准确。更好地控制了药品质量,保证人民用药安全。
[1]国家药典委员会.中华人民共和国药典(一部)[S].北京:化学工业出版社,2005.
[2]徐国钧,等.中药材粉末显微鉴别[M].北京:人民卫生出版社,1986.
[3]孙文基,谢世昌,等.天然药物成分定量分析[M].北京:中国医药科技出版社,2002.8.
[4]南京中医药大学.中药大辞典(第二版)[S].上海:上海科学技术出版社,2006.
[5]内蒙古自治区食品药品监督管理局.内蒙古蒙药制剂规范(第一册)[S].内蒙古:内蒙古人民出版社,2007.
R 291.2
A
1006-6810(2013)04-0052-03
2013年2月2日收稿
猜你喜欢
中国药学药品知识仓库(2022年13期)2022-07-03
药学实践杂志(2021年6期)2021-12-04
药品评价(2021年17期)2021-11-06
生物学通报(2021年4期)2021-03-16
化工管理(2020年18期)2020-07-15
发明与创新·小学生(2019年4期)2019-04-19
家庭医学(2014年6期)2014-09-11
药学研究(2012年2期)2012-10-25
中国医药生物技术(2012年5期)2012-02-03
