
钙钛矿型La1-xCaxCoO3纳米孔材料在Al-H2O2半燃料电池中的应用
2012-12-21 06:32庄树新刘素琴张金宝涂飞跃黄红霞黄可龙李艳华
物理化学学报 2012年2期
庄树新 刘素琴,* 张金宝 涂飞跃 黄红霞 黄可龙 李艳华
(1中南大学化学化工学院,长沙410083;2桂林科技大学化学与生物工程学院,广西桂林541004)
钙钛矿型La1-xCaxCoO3纳米孔材料在Al-H2O2半燃料电池中的应用
庄树新1刘素琴1,*张金宝1涂飞跃1黄红霞2黄可龙1李艳华1
(1中南大学化学化工学院,长沙410083;2桂林科技大学化学与生物工程学院,广西桂林541004)
通过改进的无定形柠檬酸前驱体法制备钙钛矿型La1-xCaxCoO3(x=0.2,0.4,0.5)系列化合物.使用循环伏安法和恒电流测试La1-xCaxCoO3系列化合物对于过氧化氢的电催化还原性能.同时也检测了La1-xCaxCoO3化合物中La与Ca的摩尔比及煅烧温度对其催化性能的影响.在La1-xCaxCoO3系列化合物中,650°C煅烧的La0.6Ca0.4CoO3展现出最佳的催化活性.在含有0.4 mol·dm-3H2O2的3.0 mol·dm-3KOH水溶液中,使用这种材料作为铝-H2O2半燃料电池的阴极催化剂,在150 mA·cm-2电流密度下该电池的电压为1.34 V,能量密度为201 mW·cm-2.
铝半燃料电池;钙钛矿;过氧化氢电还原;碱性介质
1 Introduction
Hydrogen peroxide(H2O2)has been investigated as an attractive oxidant instead of oxygen for several types of liquid-based fuel cells.Compared to oxygen,H2O2is liquid,resulting in an easier handling and storage.Moreover,electroreduction kinetics of H2O2is faster than that of oxygen.Therefore,fuel cells with H2O2as an oxidant tend to have better performance and are more compact than those using oxygen.The application of H2O2in several types of fuel cells has been widely studied,including direct methanol-H2O2fuel cells,1-3borohydride-H2O2fu-el cells,4-9and metal-H2O2semi fuel cells.10-16Research results showed that these fuel cells exhibited high energy density, good performance,and workability without air.
Among various electrocatalysts for H2O2reductions,such as noble metals,17-23macrocycle complexes of transition metals,24-26and transition metal oxides,27-29noble metals are usually known to be fairly active for H2O2electroreduction.Noble metal catalysts,however,have some serious problems to be overcome before worldwide spread of fuel cells,such as high cost and limited resources.Although macrocycle complex of transition metals is another considerable candidate as H2O2electroreduction catalyst showing high reduction potential and no excessive oxygen evolution,30it requires specific environments,specific temperature ranges,and are less stable than conventional noble metal catalysts.
Perovskite-type oxides which have the general formula ABO3, especially La1-xCaxCoO3,as the promising bifunctional catalyst of air electrodes have been extensively investigated.31-33And they can be found in many demonstration batteries.34,35Their catalytic activity,ionic and electronic conductivities can be tuned by partially replacing the elements in either A-site(such as La and Ca)or B-site (such as Co,Mn,Ni,Fe,and Cu).More recently,the electrocatalytic behavior of La0.6Ca0.4CoO3towards oxygen reduction in an ambient temperature alkaline electrolyte has been reported by our group,36,37finding that La0.6Ca0.4CoO3exhibited good catalytic activity for oxygen reduction.Hence,it will be interesting to study the catalytic behavior of perovskite-type La1-xCaxCoO3on H2O2reduction.
The aim of this work is to open up the possibility for development of perovskite-type compounds as H2O2cathode.Nanoporous perovskite La1-xCaxCoO3were synthesized by a modified amorphous citrate precursor(ACP)method.The electrocatalytic activity and stability of La1-xCaxCoO3for H2O2reduction in alkaline medium were investigated by cyclic voltammetry and chronoamperometry.The performance of La0.6Ca0.4CoO3as the cathode catalyst of an alkaline aluminun-H2O2semi fuel cell was evaluated.And the electrochemical performance of Al-H2O2semi fuel cell using nanoporous La1-xCaxCoO3cathode was investigated.
2 Experimental
2.1 Preparation and characterization of La1-xCaxCoO3
The catalyst La1-xCaxCoO3powder was synthesized by a modified-ACP method.37Carbon black(Vulcan XC-72R,30 nm, Cabot)was used as a pore-forming material.La(NO3)3·6H2O, Ca(NO3)2·4H2O,Co(NO3)3·6H2O,and citric acid(Sinopharm Chemical Reagent Co.,Ltd,China)used as reaction reagents were all analytical grade.An aqueous solution of citric acid with a 10% (molar fraction)excess over the number of ionic equivalents of cations was prepared.The aqueous solution of citric acid was then added by a fixed amount of carbon black,in which the mass ratio of carbon black to the theoretical mass of La1-xCaxCoO3was 3:1. Then they were agitated vigorously for 2 h in room temperature. Subsequently,the aqueous solutions of the stoichiometric metal nitrates were gradually added to the as-prepared solution and they were agitated for another 15 min.The resulting solution was concentrated by slowly evaporating water under vacuum in a rotavapor at 75°C until a gel was obtained.This gel was dried in an oven,in which temperature slowly increased to 200°C and was kept for about 12 h to produce a solid amorphous citrate precursor. The precursor was milled and calcined in air at 550,600,650,and 700°C for 4 h,respectively.
Phase identifications of the synthesized powders were conducted by a MXPAHF X-ray diffractometer(XRD)(Make Corporation,Japan)from 20°to 80°with a Cu Kαof 0.154056 nm. Thermo-gravimetry and differentialscanning calorimetry (TG-DSC)wereperformedwithaNetzschSTA 499C (NETZSCH-Gerätebau GmbH.Selb,Germany)in a flow of air (40 cm3·min-1)at a heating rate of 10°C·min-1from room temperature up to 900°C.Both the morphologies and energy dispersive X-ray spectroscopy (EDS)of the as-prepared La1-xCaxCoO3particles were observed by a Hitachi S-4800 scanning electron microscope(SEM)equipment(HITACHI,Japan).Specific surface area was measured by nitrogen adsorption-desorption with the Brunauer-Emmett-Teller(BET)method(Autosorb®-iQ,Quantachrome,America).
2.2 Preparation of La1-xCaxCoO3electrodes
To prepare La1-xCaxCoO3electrodes,La1-xCaxCoO3powder and carbon black(Vulcan XC-72)were dispersed in anhydrous ethanol by sonication for 10 min to obtain a suspension,to which a polytetrafluorethylene(PTFE,10%(w)in H2O)emulsion was added.This mixture was blended for another 30 min with ultrasonic agitation and then dried at 80°C to obtain a dough-like paste,which was finally rolled into a thin layer of about 200 μm in thickness.These thin layers had the same geometrical area of 10 mm×10 mm and the catalyst loading was approximately kept at 0.2 mg·cm-2.Two pieces of the identical thin layers were finally pressed together with a nickel foam current collector under 10 MPa pressure for 30 s and then sintered at 340°C for 30 min.The obtained electrodes consisted of 75%(mass fraction,the same below)La1-xCaxCoO3,15%carbon black,and 10%PTFE.
2.3 Electrochemical measurements
Electrochemical measurements were performed in a standard three-electrode electrochemical cell with saturated Ag/AgCl reference electrode and a bright platinum(2 cm2)foil counter electrode.The electrolyte was 3.0 mol·dm-3KOH containing H2O2with different concentrations.Cyclic voltammetry was conducted by Parstat 2273.And galvanostatic profiles were measured at various current densities for 1800 s.The reported current densities were calculated using the geometrical area of the electrode.All the solutions were made with analytical grade chemical reagents and ultra-pure water(Milli-Q,18 MΩ· cm).All potentials were referred to the saturated Ag/AgCl, KCl reference electrode.
The performance of La0.6Ca0.4CoO3as a cathode catalyst of the Al-H2O2semi fuel cell was examined by using a home-made flow through test cell as shown in Fig.1.Both the aluminum alloy anode(LF6,91.9%Al,6.5%Mg,0.6%Mn,0.3% Fe,0.3%Si,0.2%Zn,0.1%Ti)and the La0.6Ca0.4CoO3cathode have the same geometrical area of 20 mm×20 mm.Nafion 1135(DuPont)membrane was used to separate the anode and the cathode compartments.The anolyte(3.0 mol·dm-3KOH) and the catholyte(3.0 mol·dm-3KOH+0.6 mol·dm-3H2O2) were pumped into the bottom of the anode and the cathode compartments,respectively,using an individual peristaltic pump,at a flow rate of 80 cm3·min-1,and exited at the top of the compartments.The electrochemical performance of the Al-H2O2was recorded at ambient temperature on a battery test instrument(Wuhan Land Electronic Co.Ltd.,China).
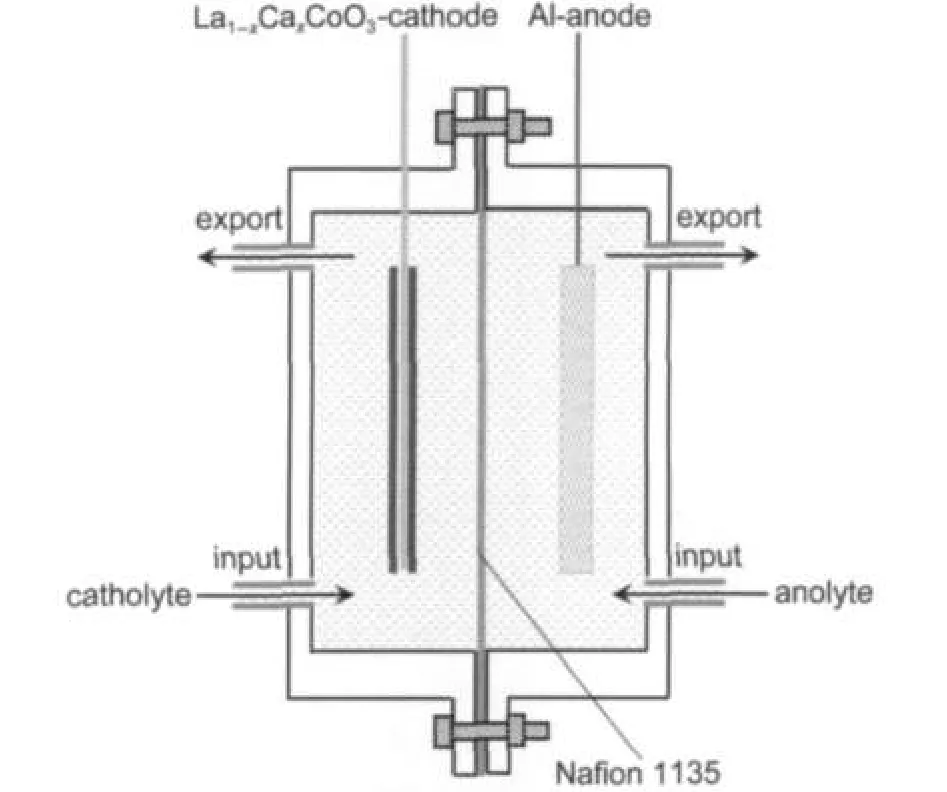
Fig.1 Schematic diagram of the plexi-glass cell used for electrochemical measurementsThe dimensions of the cell are 30 mm(long)×40 mm(wide)×40 mm(height), and the distances from the electrodes to the membrane are both 5 mm.
3 Results and discussion
3.1 Characterization of La1-xCaxCoO3
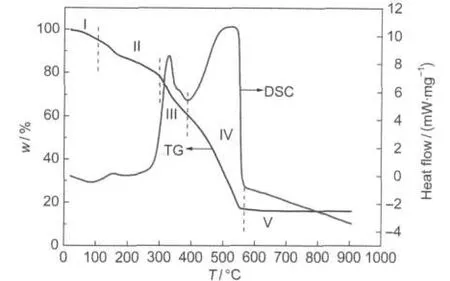
Fig.2 TG-DSC curves of the precursors prepared by the modifiedACPmethod
In order to understand the decomposition behavior and the phase evolution of the La1-xCaxCoO3precursors,the TG-DSC of the as-prepared La0.6Ca0.4CoO3precursor was conducted and the result is shown in Fig.2.The TG and DSC curves show that the whole thermal treatment process could be divided into five stages marked as stages I,II,III,IV,and V on the graph.A mass loss of 9.5%occurring in stage I(around 20-150°C)results from the removal of the residual adsorbed and hydrated water.Amass loss of 12.4%in stage II(around 150-300°C)is due to the loss of nitrate and acetic anions via decomposition and oxidation of precursors. Stage III(around 300-418°C)with a mass loss of 22.7%can be assigned to the combustion of metal citrates because the decomposition of metal citrates starts at around 320°C.38A mass loss of 38.4%in stage IV(around 418-560°C)is accompanied by a strong sharp exothermal peak at 519°C,which is attributed to the combustion of the pore-forming carbon black.39At stage V (around 560-900°C),no significant mass loss is observed,implying that almost all the nitrate anion and organic derivatives are removed and that the La0.6Ca0.4CoO3perovskite phase has already formed.
XRD analysis was performed to obtain information about the formation of crystallographic phase during thermal treatment.Fig.3 shows the XRD patterns of the La0.6Ca0.4CoO3calcined at 550,600,650,and 700°C,respectively.For the sample calcined at 550°C,the peaks corresponding to perovskite La0.6Ca0.4CoO3are observed but not so sharp,indicating that perovskite-type oxide La0.6Ca0.4CoO3has already formed but not well crystallized.The samples calcined above 600°C present a pure perovskite La0.6Ca0.4CoO3phase,without any impurity,which agrees well with the JCPDS standard pattern (36-1389).Moreover,the corresponding characteristic peaks become sharper and sharper with the calcination temperature increasing from 600 to 700°C,indicating a better crystalline structure.The average crystalline sizes of the calcined samples, calculated based on the Scherrer equation,are from 34 to 40 nm.
The morphology of La0.6Ca0.4CoO3sample calcined at 650°C together with the EDS spectrum of the particles in the selected region is shown in Fig.4.EDS only detected trace amount of the carbon element(Fig.4(b)),which might come from the carbon tap during the EDS test.As shown in Fig.4(a),most of the particles are in irregular shape,but presenting novel nanoporous structure with the particle size of about 50 nm.The interconnected pore channels would provide much convenience for the electrolyte and H2O2moving into the catalyst powders.40
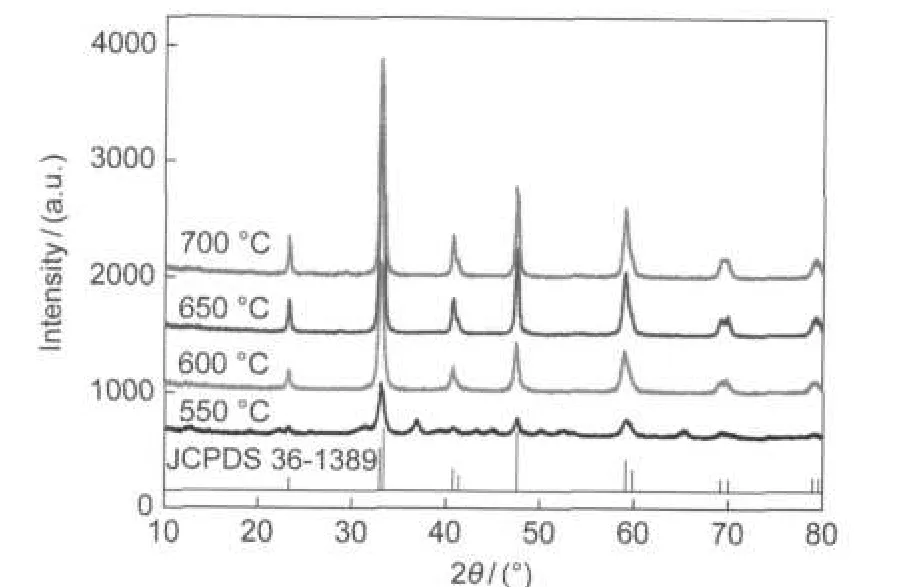
Fig.3 XRD patterns of La0.6Ca0.4CoO3calcined at different temperatures
The influence of the molar ratio of La to Ca on the crystal structure of La1-xCaxCoO3compounds was investigated by XRD spectrum.Fig.5 shows the XRD patterns of a series of La1-xCaxCoO3compounds calcined at 650°C(x=0.2,0.4,0.5). The XRD patterns are all in good agreement with corresponding JCPDS standard patterns(36-1390,36-1389,and 36-1388, respectively),indicating that this series of perovskite-type oxides La1-xCaxCoO3are all well crystallized at 650°C in the modified-ACP preparation process.In addition,the homogeneous nanoporous La1-xCaxCoO3catalysts have a high specific surface areas(205,213,215 m2·g-1for x=0.2,0.4,0.5 calcined at 650°C,respectively).It was because the carbon can not only hinder the growth of solid particles during heating41but also act as the pore-forming material after burning out which causes nanoporous structure forming when the calcination temperature reached above 550°C.Such a high specific surface area supplies a number of surface active sites,which facilitate materialʹs electrocatalytic properties.
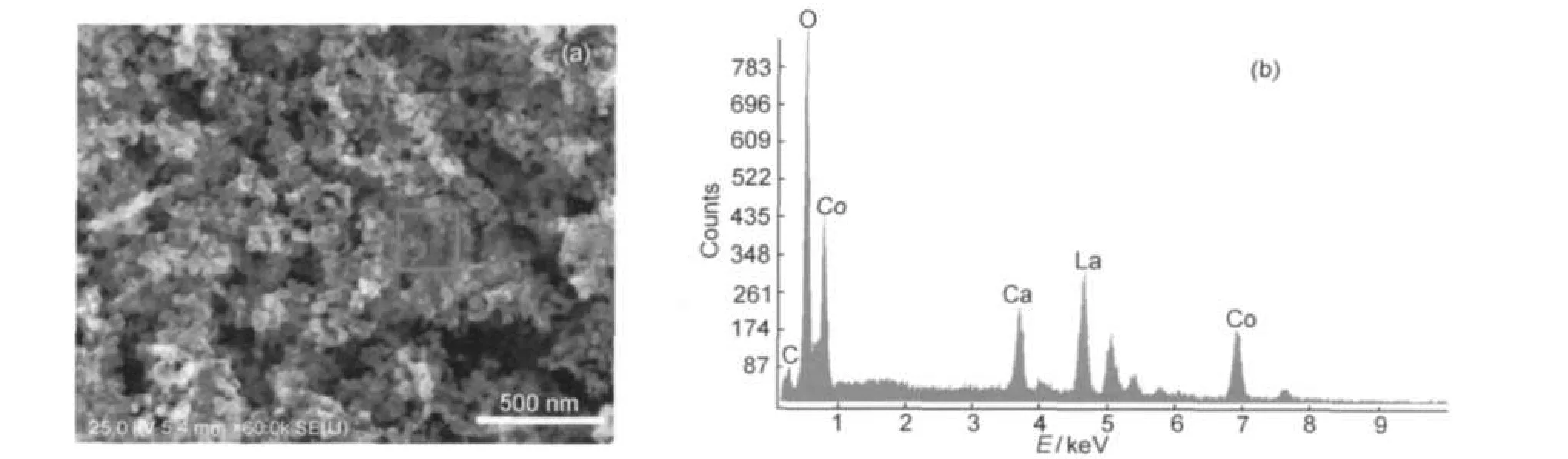
Fig.4 SEM image of La0.6Ca0.4CoO3calcined at 650°C(a)and the EDS spectra(at 15.0 kV)collected onto the square zone in the image(a)
3.2 Electrocatalytic performance of La1-xCaxCoO3
for H2O2reduction
The influence of thermal treatment temperature on the electrocatalytic performance of La0.6Ca0.4CoO3was investigated by cyclic voltammetry.The cyclic voltammograms were recorded at a scan rate of 5 mV·s-1in 3 mol·dm-3KOH containing 0.4 mol·dm-3H2O2and the results are displayed in Fig.6.The onset potential for H2O2reduction is around+0.05 V and independent of the calcination temperature.In the potential region from+0.05 to-0.2 V,the differential of current density to potential(dj/dE)increases initially and then deceases with the calcination temperature elevating from 550 to 700°C,demonstrating that the sample calcined at 650°C exhibited the highest electrocatalytic activity.This is probably because the sample calcined at 650°C achieves a better balance between crystal structure and specific surface area as indicated by TG-DSC and XRD results.Lower thermal treatment temperature favors larger specific surface area but worsens crystallinity.Higer calcination temperature leads to better crystallinity but smaller specific surface area due to the sintering.

Fig.5 XRD patterns of La1-xCaxCoO3(x=0.2,0.4,0.5) calcined at 650°C
Fig.7 shows the effect of the molar ratio of La to Ca on the electrocatalytic activity of La1-xCaxCoO3calcined at 650°C. The cyclic voltammograms were recorded at a scan rate of 5 mV·s-1in 3.0 mol·dm-3KOH containing 0.4 mol·dm-3H2O2. As shown in Fig.7,H2O2electroreduction starts at around +0.05 V,being also independent of the composition of La1-xCaxCoO3.However,the current density varies with the molar ratio of La to Ca.Among the three samples,the La0.6Ca0.4CoO3sample presents the highest dj/dE value and cathodic peak current density in the potential range from+0.05 to-0.2 V.It indicated that the sample La0.6Ca0.4CoO3exhibited the best catalytic activity for H2O2electroreduction,which was in good agreement with the references.33,42
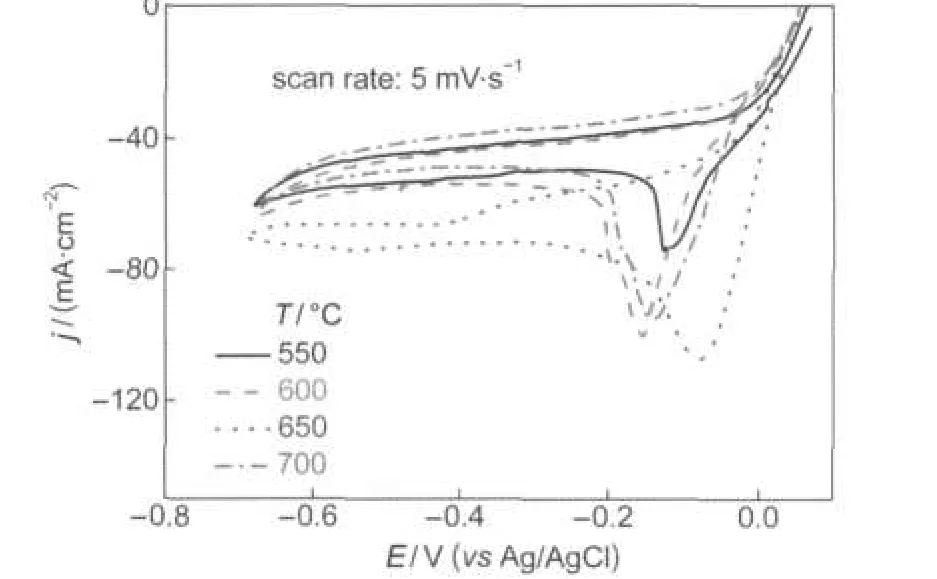
Fig.6 Cyclic voltammograms of La0.6Ca0.4CoO3calcined at different temperatures in 3.0 mol·dm-3KOH solution containing 0.4 mol·dm-3H2O2
Fig.8 presents the influence of H2O2concentration on the electroreduction activity of La0.6Ca0.4CoO3electrode.As shown in Fig.8,the dj/dE remains constant from+0.05 V to-0.2 V but the cathodic peak current density remarkably enhances with H2O2concentration increasing.The approximate linear re-lationship between the cathodic peak current density and the H2O2concentration implies that the reduction reaction was controlled by H2O2diffusion.43The nearly linear behavior of the polarization curves in the cathodic scan manifests a large ohmic resistive effect of the electrode,which is likely due to the low conductivity of La0.6Ca0.4CoO3at room temperature.Although higher limiting current density can be achieved at higher H2O2concentration, the chemical decomposition of H2O2to O2became more significant at high H2O2concentration as indicated by the formation of gas bubbles on the electrode surface.No gas bubbles were observed at H2O2concentrations below 0.4 mol·dm-3,but obvious gas evolution occurred when H2O2concentration exceeded 0.6 mol·dm-3.This indicates that the chemical decomposition of H2O2to O2becomes easier to proceed at a high H2O2concentration. Therefore,using low H2O2concentration is essential for keeping high H2O2utilization efficiency.This study demonstrated that perovskite La1-xCaxCoO3possessed considerable catalytic activity and stability towards electroreduction of H2O2in alkaline medium at room temperature.
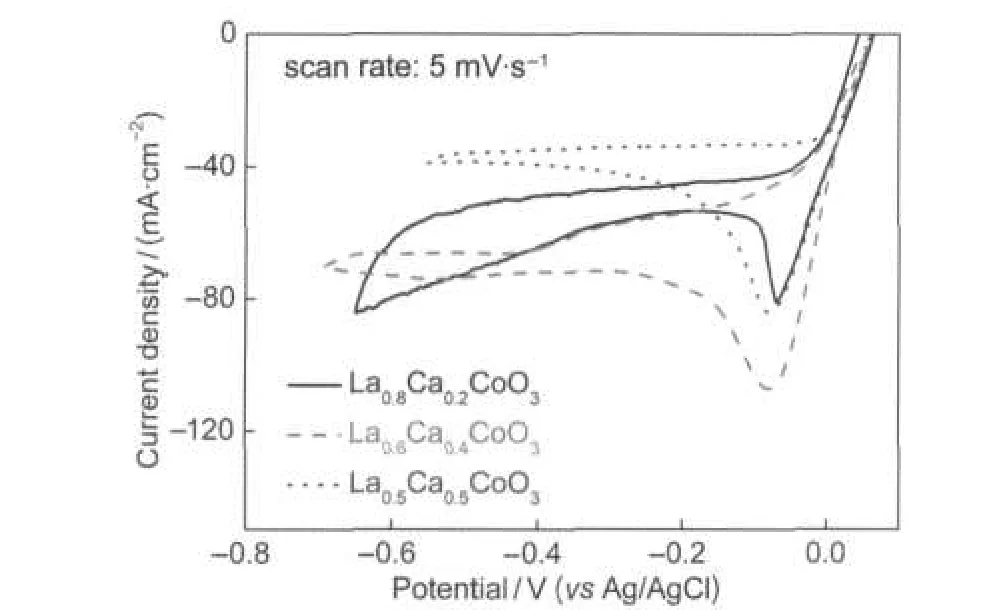
Fig.7 Cyclic voltammograms of La1-xCaxCoO3calcined at 650°C recorded in 3.0 mol·dm-3KOH containing 0.4 mol·dm-3H2O2
The stability of La0.6Ca0.4CoO3electrode for H2O2electroreduction was also investigated by galvanostatic measurements. Fig.9 shows galvanostatic curves of H2O2electroreduction on La0.6Ca0.4CoO3calcined at 650°C with various current densities.At these four current densities,the voltage agree well with the results of cyclic voltammograms.At low current densities, the voltage reaches to steady state after a few seconds and displays no sign of decrease within the test period,indicating that the catalytic performance of La0.6Ca0.4CoO3was stable and sustainable for hydrogen peroxide electroreduction.When the current density further increases(200 mA·cm-2),the voltage fluctuations are observed within 30 min test period,which is due to the depletion of H2O2near the electrode surface with 0.4 mol·dm-3H2O2.
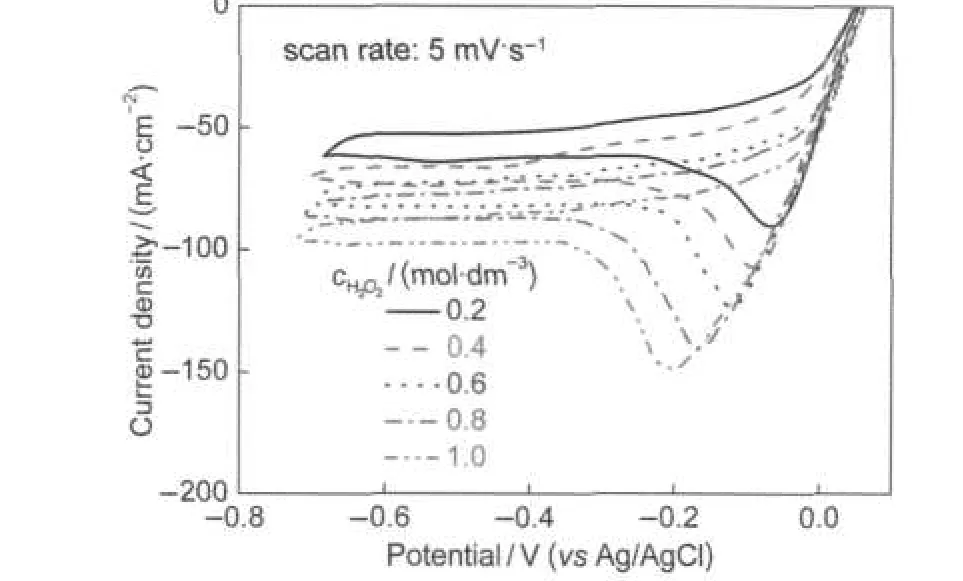
Fig.8 Cyclic voltammograms of La0.6Ca0.4CoO3measured in 3.0 mol·dm-3KOH containing H2O2with different concentrations
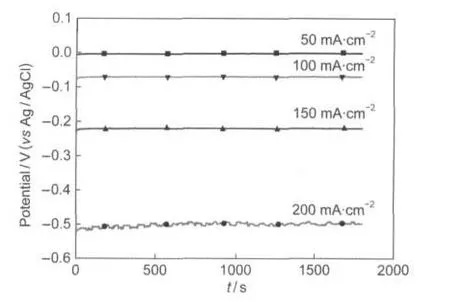
Fig.9 Galvanostatic curves for H2O2electroreduction on La0.6Ca0.4CaO3electrode at different current densities in3.0 mol·dm-3KOH+0.4 mol·dm-3H2O2
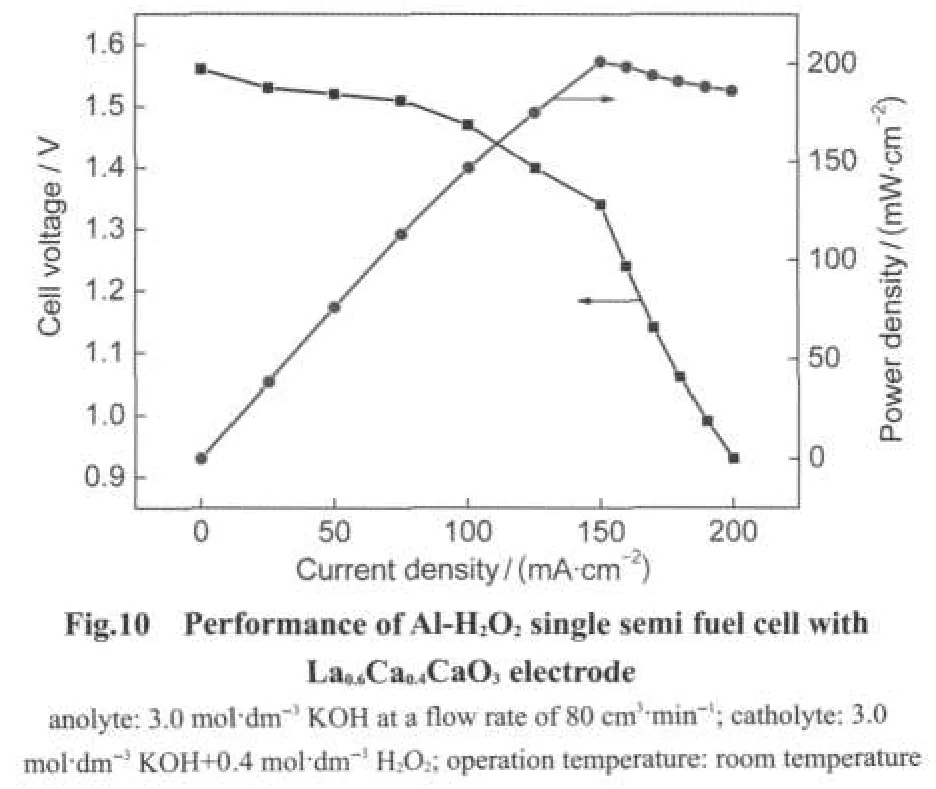
The performance of La0.6Ca0.4CaO3as the cathode catalyst of Al-H2O2semi fuel cell was investigated.Fig.10 presents plots of cell voltage and power density against current density.The cell was operated at room temperature with 0.4 mol·dm-3H2O2feeding to the cathode compartment.The cell exhibited an open circuit voltage of around 1.55 V and the cell voltage decayed almost linearly and then dropped drastically with the current density increasing.This behavior indicated that the cell performance had the poor mass transport and the large ohmic resistance,which might be attributed to the cell design.By optimizing the cell design,better cell performance might be achieved.Further investigation is undergoing in our laboratory. A peak power density of 201 mW·cm-2was obtained at 150 mA·cm-2and 1.34 V,which was comparable with that of Al-H2O2semi fuel cell using nanoparticle Co3O4cathode,27but was better than that of Al-O2semi fuel cell reported by Rota et al.44Although high power density of Al-H2O2semi fuel cell can be achieved with high concentration of H2O2,the chemical de-composition rate of H2O2at high concentration will increase the expense of H2O2.It was worth emphasizing that La1-xCaxCoO3can also be used as cathode catalysts for other types of fuel cells using H2O2as oxidant such as direct methanol-H2O2fuel cell and borohydride-H2O2fuel cell.
4 Conclusions
Perovskite-type La1-xCaxCoO3(x=0.2,0.4,0.5)compounds were prepared by a modified amorphous citrate precursor method.Based on XRD and SEM analyses,a series of compounds La1-xCaxCoO3could be synthesized at 550°C and presented homogeneous nanoporous morphology at 650°C.Their catalytic activities for H2O2electroreduction in alkaline medium were evaluated at room temperature.Dependence of catalytic activity upon the molar ratio of La to Ca in the La1-xCaxCoO3and the calcination temperature was discussed deeply.The results showed that the sample La0.6Ca0.4CaO3calcined at 650°C exhibited the best catalytic activity.The Al-H2O2fuel cell using La0.6Ca0.4CaO3as cathode catalysts displayed an open circuit voltage of 1.55 V and a peak power density of 201 mW·cm-2. Compared with precious materials such as Ru,Rh,Ir,Pt,and Pd,perovskite-type La1-xCaxCoO3are more abundant and cheaper.Hence,perovskite-type La1-xCaxCoO3are attractive electrocatalysts for H2O2electroreduction.
(1) Sung,W.;Choi,J.W.J.Power Sources 2007,172,198.
(2) Bewer,T.;Beckmann,T.;Dohle,H.;Mergel,J.;Stolten,D. J.Power Sources 2004,125,1.
(3) Prater,D.N.;Rusek,J.J.Appl.Energy 2003,74,135.
(4) Gu,L.;Luo,N.;Miley,G.H.J.Power Sources 2007,173,77.
(5) Raman,R.K.;Prashant,S.K.;Shukla,A.K.J.Power Sources 2006,162,1073.
(6) Chatenet,M.;Micoud,F.;Roche,I.;Chainet,E.;Vondrák,J. Electrochim.Acta 2006,51,5452.
(7)Pei,F.;Wang,Y.;Wang,X.;He,P.;Chen,Q.;Wang,X.;Wang, H.;Yi,L.;Guo,J.Int.J.Hydrog.Energy 2010,35,8136.
(8) Ponce de León,C.;Walsh,F.C.;Patrissi,C.J.;Medeiros,M. G.;Bessette,R.R.;Reeve,R.W.;Lakeman,J.B.;Rose,A.; Browning,D.Electrochem.Commun.2008,10,1610.
(9)Raman,R.K.;Choudhury,N.A.;Shukla,A.K.Electrochem. Solid-State Lett.2004,7,A488.
(10) Brodrecht,D.J.;Rusek,J.J.Appl.Energy 2003,74,113.
(11)Yang,W.;Yang,S.;Sun,W.;Sun,G.;Xin,Q.J.Power Sources 2006,160,1420.
(12) Popovich,N.A.;Govind,R.J.Power Sources 2002,112,36.
(13) Hasvold,Ø.;Johansen,K.H.;Mollestad,O.;Forseth,S.; Størkersen,N.J.Power Sources 1999,80,254.
(14) Hasvold,Ø.;Størkersen,N.J.;Forseth,S.;Lian,T.J.Power Sources 2006,162,935.
(15)Yang,W.;Yang,S.;Sun,W.;Sun,G.;Xin,Q.Electrochim.Acta 2006,52,9.
(16) Patrissi,C.J.;Bessette,R.R.;Kim,Y.K.;Schumacher,C.R. J.Electrochem.Soc.2008,155,B558.
(17) Savinova,E.R.;Wasle,S.;Doblhofer,K.Electrochim.Acta 1998,44,1341.
(18) Prakash,J.;Joachin,H.Electrochim.Acta 2000,45,2289.
(19) Strbac,S.Electrochim.Acta 2011,56,1597.
(20) Zeis,R.;Lei,T.;Sieradzki,K.;Snyder,J.;Erlebacher,J. J.Catal.2008,253,132.
(21)Qin,X.;Wang,H.;Wang,X.;Miao,Z.;Fang,Y.;Chen,Q.; Shao,X.Electrochim.Acta 2011,56,3170.
(22) Fu,R.;Zheng,J.S.;Wang,X.Z.;Ma,J.X.Acta Phys.-Chim. Sin.2011,27,2141.[符 蓉,郑俊生,王喜照,马建新.物理化学学报,2011,27,2141.]
(23) Wu,Y.N.;Liao,S.J.Acta Phys.-Chim.Sin.2010,26,669. [吴燕妮,廖世军.物理化学学报,2010,26,669.]
(24)Bouwkamp-Wijnoltz,A.L.;Visscher,W.R.;van Veen,J.A. Electrochim.Acta 1998,43,3141.
(25) Raman,R.K.;Shukla,A.K.J.Appl.Electrochem.2005,35, 1157.
(26) Herrmann,I.;Kramm,U.I.;Fiechter,S.;Bogdanoff,P. Electrochim.Acta 2009,54,4275.
(27) Cao,D.;Chao,J.;Sun,L.;Wang,G.J.Power Sources 2008, 179,87.
(28) Goldik,J.S.;Nesbitt,H.W.;Noël,J.J.;Shoesmith,D.W. Electrochim.Acta 2004,49,1699.
(29) Keech,P.G.;Noël,J.J.;Shoesmith,D.W.Electrochim.Acta 2008,53,5675.
(30) González,G.L.;Kahlert,H.;Scholz,F.Electrochim.Acta 2007, 52,1968.
(31)Zhang,H.M.;Shimizu,Y.;Teraoka,Y.;Miura,N.;Yamazoe,N. J.Catal.1990,121,432.
(32) Tanaka,H.;Misono,M.Curr.Opin.Solid State Mater.Sci. 2001,5,381.
(33) Weidenkaff,A.;Ebbinghaus,S.G.;Lippert,T.Chem.Mater. 2002,14,1797.
(34) Lee,C.K.;Striebel,K.A.;McLarnon,F.R.;Cairns,E.J. J.Electrochem.Soc.1997,144,3801.
(35) Müller,S.;Holzer,F.;Haas,O.J.Appl.Electrochem.1998,28, 895.
(36) Zhuang,S.;Huang,K.;Huang,H.;Liu,S.;Fan,M.J.Power Sources 2011,196,4019.
(37) Zhuang,S.;Huang,C.;Huang,K.;Hu,X.;Tu,F.;Huang,H. Electrochem.Commun.2011,13,321.
(38) Deganello,F.;Marcì,G.;Deganello,G.J.Eur.Ceram.Soc. 2009,29,439.
(39) Jakab,E.;Omastová,M.J.Anal.Appl.Pyrolysis 2005,74,204.
(40) Yang,J.;Xu,J.J.Electrochem.Commun.2003,5,306.
(41) Liu,H.P.;Wang,Z.X.;Li,X.H.;Guo,H.J.;Peng,W.J. J.Power Sources 2008,184,469.
(42)Kahoul,A.;Hammouche,A.;Poillerat,G.;De Doncker,R.W. Catal.Today 2004,89,287.
(43)Wang,G.;Bao,Y.;Tian,Y.;Xia,J.;Cao,D.J.Power Sources 2010,195,6463.
(44) Rota,M.;Comninellis,C.;Muller,S.;Holzer,F.;Haas,O. J.Appl.Electrochem.1995,25,114.
September 6,2011;Revised:November 23,2011;Published on Web:November 29,2011.
Application of Nanoporous Perovskite La1-xCaxCoO3in an Al-H2O2Semi Fuel Cell
ZHUANG Shu-Xin1LIU Su-Qin1,*ZHANG Jin-Bao1TU Fei-Yue1
HUANG Hong-Xia2HUANG Ke-Long1LI Yan-Hua1
(1College of Chemistry and Chemical Engineering,Central South University,Changsha 410083,P.R.China;
2College of Chemistry and Bioengineering,Guilin University of Technology,Guilin 541004,Guangxi Province,P.R.China)
Perovskite-type series of compounds La1-xCaxCoO3(x=0.2,0.4,0.5)were synthesized by a modified amorphous citrate precursormethod.Theircatalytic activities forhydrogen peroxide electroreduction in 3.0 mol·dm-3KOH at room temperature were evaluated by cyclic voltammetry and galvanostatic measurements.The influences of annealing temperature and the molar ratio of La to Ca of La1-xCaxCoO3on catalytic performance were investigated.Among the series of compounds,La0.6Ca0.4CoO3calcined at 650°C exhibited the highest catalytic activity.An aluminum-hydrogen peroxide semi fuel cell using La0.6Ca0.4CoO3as cathode catalyst achieved a peak power density of 201 mW·cm-2at 150 mA·cm-2and 1.34 V running in 0.4 mol·dm-3H2O2.
Aluminium semi fuel cell;Perovskite;Hydrogen peroxide electroreduction; Alkaline medium
10.3866/PKU.WHXB201111293
*Corresponding author.Email:zsxtonny@yahoo.com.cn;Tel:+86-31-88879850.
The project was supported by the National High-Tech Research and Development Program of China(863)(2008AA031205)and Graduate Degree Thesis Innovation Foundation of Central South University,China(1343-74334000005).
国家高技术研究发展计划(863)(2008AA031205)和中南大学博士创新基金(1343-74334000005)资助项目
O646
猜你喜欢
科技进步与对策(2021年12期)2021-06-23
安全(2021年4期)2021-05-19
歌海(2021年1期)2021-04-02
湖南包装(2020年6期)2021-01-20
学苑创造·A版(2018年12期)2018-03-04
物理学进展(2017年1期)2017-02-23
云南师范大学学报(自然科学版)(2015年5期)2015-12-26
文贝:比较文学与比较文化(2015年1期)2015-11-14
小主人报(2015年1期)2015-03-11
太阳能(2015年4期)2015-02-28
