
聚乙烯唑啉作用下甲烷水合物分解的分子动力学模拟
2012-12-21 06:33王燕鸿陈玉娟郎雪梅樊栓狮
物理化学学报 2012年7期
王燕鸿 陈玉娟 包 玲 郎雪梅 樊栓狮
(华南理工大学化学与化工学院,传热强化与过程节能教育部重点实验室,广州510640)
聚乙烯唑啉作用下甲烷水合物分解的分子动力学模拟
王燕鸿 陈玉娟 包 玲 郎雪梅 樊栓狮*
(华南理工大学化学与化工学院,传热强化与过程节能教育部重点实验室,广州510640)
利用分子动力学模拟系统研究了不同质量浓度下(1.25%、2.50%、6.06%)聚乙烯唑啉(PEtO)对甲烷水合物的分解作用.模拟体系为甲烷水合物2×2×2的超胞和聚合物对接体系.模拟发现水分子间氢键构架的水合物笼型结构在PEtO的作用下出现扭曲,最终导致水合物笼型结构完全坍塌.通过氧原子径向分布函数、均方位移以及扩散系数比较不同浓度PEtO的作用,证实在一定浓度范围内,PEtO的浓度越高,其水合物分解作用越强.此外,PEtO具有一定的可生物降解性.PEtO对水合物的作用为:PEtO吸附在水合物表面,其中的酰胺基(N―C=O)与成笼的水分子形成氢键,破坏邻近的笼形结构,令水合物分解;PEtO不断分解表面的水合物,直到水合物笼完全分解.
甲烷水合物;聚乙烯唑啉;分子动力学模拟;水合物抑制剂
1 Introduction
Gas clathrate hydrates are crystalline inclusion compounds which are composed of gas and water.1Water molecules form a lattice structure with cavities through hydrogen bonds by van der Waals type dispersion force.The cavities are occupied by gas molecules,such as methane,ethane,propane etc.at high-pressure and low-temperature.2
Since discovered by Hammerschmidt in 1934,3gas hydrate has attracted much attention as a major potential threat in oil and gas transportation.Kinds of methods have been attempted to prevent hydrate blockages.The most available way is to use thermal hydrate inhibitors,such as methanol,glycol,etc.Thermal hydrate inhibitors prevent hydrate form by shifting the hydrate phase boundary to lower temperature and higher pressure.However,high concentrations(up to 60%(w,mass fraction))of inhibitor required for certain field cases lead to high cost and environmental pollution.Kinetic hydrate inhibitors (KHIs),acting effectively at low concentration,had aroused much attention.
Currently available KHIs work at a subcooling of 13°C, which still does not satisfy many applications of the oil and gas industry.Developing new KHIs to act at higher subcooling or lower concentration is a great interest.How inhibitors act and affect the formation and growth of hydrate is crucial for the designing of new inhibitors,whereas it is still poorly known. Many researchers have carried out relevant study,employing molecular dynamics(MD)simulation on mechanism of hydrate inhibitor.
Rodgerʹsgroup4studied the inhibition mechanism of poly(N-vinyl pyrrolidone)(PVP)to{111}face of type II clathrate hydrate by MD in 1996,and they found that the monomeric unit was adsorbed onto the surface and the adsorption site was determined by the location of the pendant hydrogen.In 1999,they5continued to investigate the structural behavior of PVP monomer and liquid water.Synergistic solvation effects arising from the conjunction of hydrophobic solvation(around the methyl group)and hydrophilic solvation(around the amide oxygen)might play an important role in the structuring effect of the pyrrolidone ring on surrounding water.In 2004,they6designed a new kind of KHI-tributylammoniumpropylsulfonate (TBAPS)and used MD to study its performance.TBAPS,with sulfonate-induced structure to propagate strongly over several solvation shells,had an activity comparable with PVP.In 2006,they7worked on poly(dimethylaminoethylmethacrylate) (PDMAEMA)to determine the effect of polydispersity in a real polymeric inhibitor.They8carried out the MD simulation the next year to study the inhibition mechanism of PVP and PDMAEMA of the nucleation process for methane hydrate and put forward the mechanism of surface energy which was very different from previous surface adsorption and structuring effect mechanism.The following year they investigated PMAEMAʹs inhibition performance to water/methane system at different temperatures to demonstrate the impact of temperature.9
Kvammeʹs groupdid MD simulations of PVP kinetic inhibitor in liquid water and hydrate/liquid water systems in 1997. In 2005,they11researched poly(N-vinyl caprolactam(PVCap) and confirmed PVCap outperformed PVP as a kinetic hydrate inhibitor using MD.In 2010,they12carried out a study to determine the relation of the inhibition characteristic of PVP and PVCap polymers with molecular weight,and supposed that there would exist an inhibitor layer to hinder the diffusion of hydrate formers.
Wan et al.13studied methane hydrate dissociation process in the presence of thermodynamic inhibitor—ethylene glycol by MD simulation.Results indicated that hydrate dissociation started from the surface layer of the solid hydrate and then gradually expanded to the internal layer.
Balbuena et al.14studied the behavior and activity of PVP, PVCap etc.in clathrate-methane-water system and found that the hydrophobic inhibitors might block the surface of the nascent crystals,whereas the hydrophilic ones disrupting the water structure mainly happened at initial stages of clathrate formation.
Andersonʹs group15,16considered that the inhibition mechanism of KHI can be stated as the following 2 steps:(1)inhibitor molecules disrupt the local organization of the water and guest molecules,increasing the barrier to nucleation and nuclei propagation;(2)once nucleation occurs,the inhibitor binds to the surface of hydrate nanocrystal and retards growth along the bound growth plane.
Rodgerʹs simulation results8showed that PVP induced dissolution of small hydrate clusters without any direct contact between the additive and the hydrate surface.They supposed that PVP increased the interfacial surface energy without adsorbing onto hydrate surface,thus disputing step(2)of Andersonʹs hypothesis.Kvammeʹs group12agreed in general with step(1)of Anderson hypothsis,while assumed that PVP and similar low dosage hydrate inhibitor(LDHI)would form a layer between the water and the gas phase and affect the diffusion of hydrate former.
KHIs have been in commercial and used in oil and gas industry for about 18 years.Among these commercial KHIs,most of them contain amide groups.Exxon Company17deduced that a key structure in many KHI polymers was an amide group attached to a hydrophobic group in repeating unit.Though a number of experiments have tested that polymer had good kinetic inhibition,there were few direct facts to prove that aminogroup played a main role in KHI inhibitors.Present work is focused on the amino-group to investigate the CH4hydrate decomposing process in the presence of amide groups by dynamics simulation.
Poly(2-ethyl-2-oxazoline)(PEtO)is a nontoxic,biodegradability polymer.Karaaslan and Parlaktuna18have already tested PEtO at 1%concentration and find that it can inhibit CH4hydrate formation at high pressures and low temperatures.Without the regular ring structure of KHIs,PEtO has simple struc-ture,only containing amide groups in repeating unit.To avoid the effect of ring structure,we chose PEtO as KHI and investigated the inhibition effect of amide group in this work by dynamics simulation.
2 Simulation method
2.1 Model details
CH4hydrate unit cell is a crystal with a lattice parameter of 1.189 nm belonging to the cubic space group Pm3n.There are 46 water molecules and 8 CH4molecules in type I CH4hydrate unit cell at 100%occupancy.The simulation layer system consisted of a CH4hydrate with 2×2×2 supercell crystal and kinetic inhibitor PEtO at different concentrations.
Oxygen positions were set according to X-ray diffraction experiments.19-21Hydrogen atoms were placed in a manner consistent with the 2-fold crystallographic texture,following the Bernal-Fowler rule.Water molecules were modeled using the SPC potential(partial point charge of O and H atom:-0.82e, 0.41e,respectively)and methane with united atom potentials. Lennard-Jones interactions were used between O atoms to account for short-range repulsion and long-range dispersion interactions between water molecules.Potential parameters of CH4hydrate were given an accurate description in Rodgerʹs previous studies.4-6The PEtO was modeled using an all-atom CHARMm force filed.An orthorhombic supercell hydrate was constructed based on type I hydrate unit cell.There were 368 water molecules and 64 CH4gas molecules in the hydrate system with a cell parameters of 2.378 nm×2.378 nm×2.378 nm (x×y×z)(see Fig.1).The most potential chemical structures as inhibitors often possess active groups for attaching themselves to the hydrate surface including the double-bond oxygen,the hydroxyl groups,and the nitrogen.22Fig.2 shows the PEtO unit structure,which contains double-bond oxygen and nitrogen. PEtO at concentrations of 1.25%,2.50%,6.06%with molecule numbers of 1,2,5,respectively were added to study the decomposition effect to CH4hydrate.All atomic positions were allowed to freely fluctuate during the simulation.
2.2 Simulation details

Fig.1 A2×2×2 super cell of structure I CH4hydrateball-stick for H2O molecules;stick for CH4molecules;dashed lines for the hydrogen bonding network between H2O molecules

Fig.2 Structure of poly(2-ethyl-2-oxazoline)(PEtO)
The molecular dynamics simulations were performed based on Materials Studio.23When the CH4hydrate-PEtO system was built,optimization was carried out using the steepest descent and conjugate gradient method.19The final configuration of layer system was acquired after geometry and energy optimization.MD simulation calculations were conducted using NVT ensemble.Periodic boundary conditions were applied.Consistent valence force field(CVFF)was employed to conduct the mutual forces between CH4gas molecules.Long-range Coulomb forces were treated by Ewald.24The water parameters were as follows:O―H bond length was 0.096 nm,H―O―H bond angle was 104.520°.Temperature of systems was kept at 273.15 K by Nose-Hoover thermostat.25The time step was set as 1 fs and total simulation time was 3000 ps.
3 Results and discussion
After the CH4hydrate-PEtO layer system was conducted in NVT ensemble,the trajectory of system and final conformation were available.Several methods have been employed to investigate the influence of inhibitors interacted with the CH4hydrate.Great attention has been paid to structural effect of PEtO on hydrate including the radial distribution functions(RDFs), and configurations of layer system.Results on dynamics effect like mean square displacement of oxygen atoms,diffusion coefficient of water molecules were also investigated.
3.1 Snapshots of the layer system configurations
Snapshots of configurations of the dynamics simulation layer system at different PEtO concentrations at 273.15 K are presented in Fig.3-Fig.7.Fig.3 shows that all layer structures are of perfect CH4hydrate,informed by the neat hydrogen bond network between H2O molecules without distortions at the initial time.As shown in Fig.4,an obvious distortion of the hydrogen bond in hydrate cages appears after 1 ps.Some CH4molecules have escaped from the original cages and aggregated together,demonstrating that the decomposition of hydrate has started at the interface,which is considered as the best mass transmission and heat transfer place.The surface of hydrate crystal is distorted and CH4molecules escape from the cages at edge.Nevertheless,cages in the bulk of hydrate crystal are still not affected by inhibitor polymer.
The change process of hydrate from 50 to 100 ps are presented Fig.5 and Fig.6,showing that hydrate cages is out of shape in the presence of PEtO.The ellipsoid parts are given in detail to show how PEtO interacts with water molecules.Dissolved at the interface,PEtO gets close and permeates into the hydrate crystal which benefits mass transmission.It is observed that the double-bonded oxygen atoms of polymer combine with hy-drogen atoms and form hydrogen bonds shown in Fig.5 and the detailed figures,both at concentrations of 2.50%and 6.06%. Hydrogen bonds between water molecules have been disrupted by“foreign hydrogen”,which conforms to previous studies,26,27Double-bonded oxygen is likely to form hydrogen bonds with hydrogen atoms at appropriate conditions and may form more than one hydrogen bond.Once the original hydrogen bonds are disrupted or displaced,the hydrate cage turns to be imperfect with the trapped CH4coming out,i.e.,a decomposition of the touched hydrate cages.As simulation going on,by the time of 100 ps(Fig.6),PEtO penetrates into the bulk and double-bonded oxygen of group N―C=O combines with hydrogen atoms of water molecules to form hydrogen bonds and distort hydrogen bond in hydrate cages.The hydrophilic groups produce steric hindrance preventing the decomposing water molecules from regenerating hydrogen bonds with each other,inhibiting the growth of hydrate.

Fig.3 Initial configurations of CH4hydrate crystal-PEtO layer systemswPEtO/%:(a)1.25,(b)2.50,(c)6.06;the big ball-stick for PEtO molecule with a darker color to sign oxygen atoms;the small ball-stick for H2O molecules; stick for CH4molecules;dashed lines for the hydrogen bonding network between H2O molecules
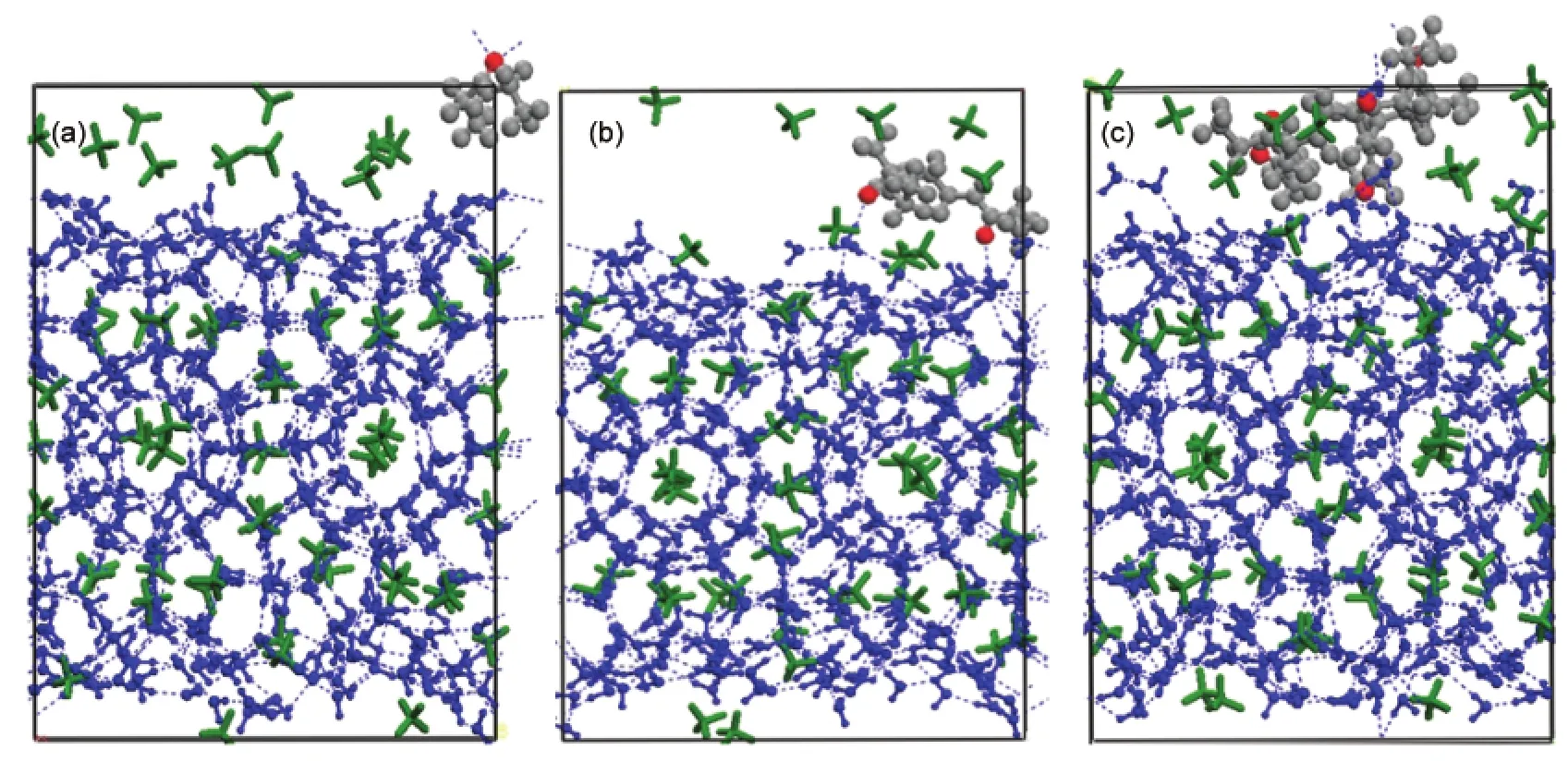
Fig.4 Snapshot configurations of CH4hydrate crystal-PEtO layer systems at 1 pswPEtO/%:(a)1.25,(b)2.50,(c)6.06
Fig.7 presents final configurations of CH4hydrate-PEtO layer systems(3000 ps)with three concentrations.Evident collapse of the hydrogen bonding network in CH4hydrate can be observed.Inhibitor polymer has pierced the layer system.Hydrogen bonds and cages in original perfect crystal are in a complete chaos status.Almost all CH4molecules have run away from the initial positions and aggregate together at the interface,but do not disperse in liquid water because CH4is slightly soluble in water.The final configurations of CH4hydrate crystal-PEtO layer systems with three concentrations are completely collapsed and have few differences.
3.2 Radial distribution function of oxygen atoms
Radial distribution function gαβ(r)is an important parameter to identify the degree order of solid or liquid structures,like the ratio of the area density to the average density.RDF shows the probability of finding the same kinds of atoms from central atom,whose expression is

where Nαis total amount of α particles;Nβis total amount of β particles;Vsis the volume of the system;niβ(r)is amount of β
particles in the area of r to(r+Δr).

Fig.5 Snapshot configurations of CH4hydrate crystal-PEtO layer systems at 50 pswPEtO/%:(a)1.25,(b)2.50,(c)6.06.The below are detailed figures of ellipsoid parts to show how oxygen atoms of PEtO interact with water molecules.

Fig.6 Snapshot configurations of CH4hydrate crystal-PEtO layer systems at 100 pswPEtO/%:(a)1.25,(b)2.50,(c)6.06.The below are detailed figures of ellipse parts to show how oxygen atoms of PEtO interact with water molecules.
There are two typical peaks at distances of 0.27 and 0.44 nm in RDFs for hydrate crystals,while the second characteristic peak disappears in liquid water.RDFs of oxygen atoms of water in three different CH4hydrate systems at 273.15 K are presented in Fig.8.Such figures display that the RDFs of oxygen atoms gOO(r)fluctuate within simulation time,showing great differences between the inhibited and uninhibited(0 ps)hydrates.As presented in Fig.8(a),gOO(r)of the first peak(0.27 nm)in gOO(r)of water molecules is 24.15,gOO(r)of the second peak(0.44 nm)is 5.03 at the beginning of dynamics simulation,indicating the initial structure is perfect hydrate crystal. The first peak decreases from 24.15 to 5.56,and the second peak also reveals a significant declining trend after 1 ps.At 50 ps,the peaks height decreases continuously.The profiles vary more violently and the second peak almost disappears at 100 ps.The gOO(r)at 100 and 3000 ps are nearly of the same,peak at 0.44 nm gone.RDF characteristic peak height decreases because“foreign molecules”(PEtO)interact with water molecules in CH4hydrate,changing the network structure with hydrogen between water molecules in hydrate.With the impact of PEtO at concentration of 1.25%,perfect CH4hydrate system becomes a disorder.Furthermore,the peaks decline faster and in a larger scale,CH4hydrate is in a more disorder state.RDFs of O atoms at 2.50%and 6.06%PEtO(Fig.8(b)and Fig.8(c)) show the same trends as that of 1.25%.When the simulation goes on for 50 ps,the first and the second peaks have fallen to a large extend,especially for the 6.06%PEtO.At the time of 100 ps,the two basic characteristic peaks of hydrate almost vanish completely,demonstrating that solid water has already changed to liquid water.PEtO shows the ability in decomposing CH4hydrate.

Fig.7 Snapshots of final configurations(at 3000 ps)of CH4hydrate crystal-PEtO layer systemswPEtO/%:(a)1.25,(b)2.50,(c)6.06
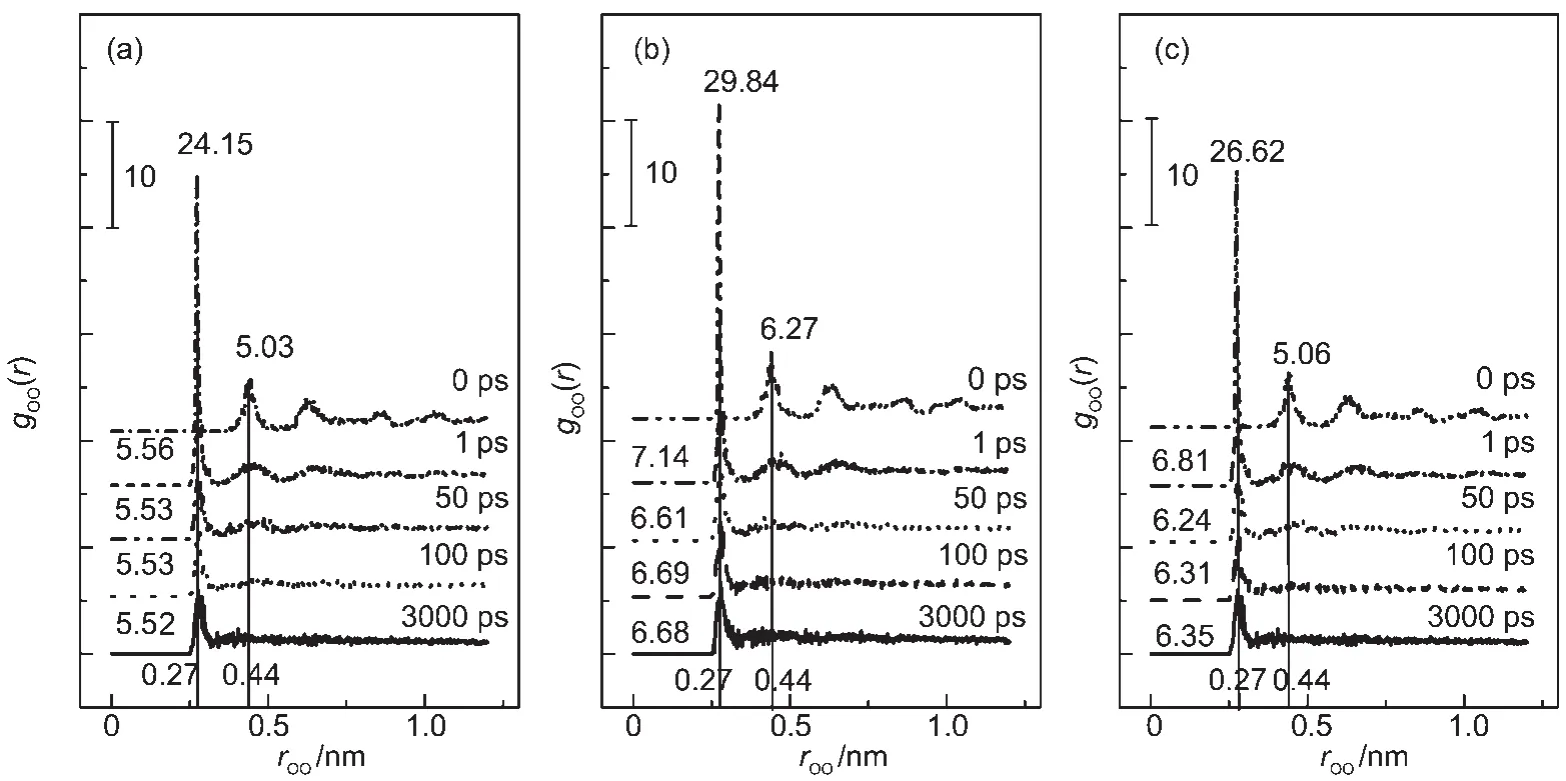
Fig.8 Radial distribution functions of oxygen atoms of CH4hydrate at different simulation time with PEtO concentrations of 1.25%(a),2.5%(b),and 6.06%(c)
3.3 Mean square displacement(MSD)of oxygen
atoms
Mean square displacement is a measure of the average distance of a molecule travels.MSD is on the behalf of movements away from equilibrium position of atoms within the whole simulation process due to inter-molecule forces,indicating the intensity between molecules.The expression of MSD defines as follows:
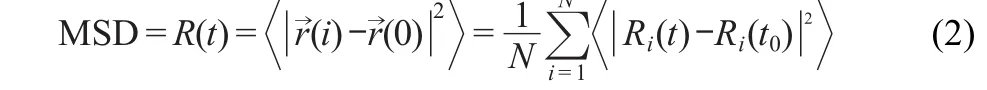
where R(t)represents the mean square displacement of the atoms during the simulation time;Ri(t0)is atom i position at the time of t0;N is the amount of the total atoms.

Fig.9 MSD of oxygen atoms in CH4hydrate with PEtO
MSD can be used to identify whether the simulation system is in solid or in fluid.As for a perfect crystal,the constituent molecules vibrate around their lattice sites.Regarding to solid hydrate crystal,all water molecules are in a relatively fixed lattice point,and can not deviate far away from the lattice point, and MSD variation curve with time is a line parallel to X axis. As for liquid water,water molecules are in disorder.The MSD variation curve with time will increase with time going on.
The MSD curves of oxygen atoms of CH4hydrate in the presence of three PEtO concentrations(1.25%,2.50%,6.06%) are shown in Fig.9.The MSD of oxygen atoms increases greatly with time at three concentrations and declares that PEtO has a good effect on decomposing CH4hydrate.Besides, it is observed that the MSD of oxygen atoms at a concentration of 1.25%is smaller than those of 2.50%and 6.06%.With PEtO concentration increasing,its inhibition effect gets stronger.But when PEtO concentrations exceed 2.50%,inhibition effect increases little.The inhibition effect of 6.06%PEtO is closed to that of 2.50%PEtO.In conjunction with previous studies,28it is conformed that PEtO is a low dosage hydrate inhibitor,which can be as active as PVP even at 1%.
3.4 Diffusion coefficient of water molecules
Based on statistics,the diffusion coefficient(D)of dynamics simulation system can be defined as follows:
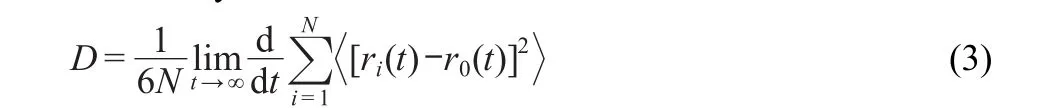
The diffusion coefficient is closely related to mean square displacement.If a straight line is fitted for the mean square displacement curve as:y=ax+b,the diffusion coefficient is deduced as D=a/6.The diffusion coefficient of water molecules is calculated based on the above-mentioned formula.
Chemical reaction is an interaction between different molecules with mass and heat transfer.The higher diffusion coefficient of water molecules means higher heat and mass transmission.Decomposition of CH4hydrate is an endothermic process. It is helpful to deliver or absorb heat for the CH4hydrate system decomposition.Higher diffusion coefficient makes hydrate inhibitor contact sufficiently with CH4hydrate easier and faster,and results in a good decomposing effect on CH4hydrate.
The diffusion coefficient of water molecules in CH4hydrate with PEtO concentration at 1.25%is 1.34×10-9m2·s-1.Compared to 1.34×10-9m2·s-1,the values of concentrations at 2.50%(1.50×10-9m2·s-1)and 6.06%(1.52×10-9m2·s-1)have increased by 11.9%and 13.4%,respectively.Thus,it suggests that the decomposing effect of 2.50%PEtO is similar to that of 6.06%PEtO,and both greater than that of 1.25%.
The decomposing effect of PEtO on CH4hydrate is derived from chemical structure and functional group.PEtO contains a hydrophilic functional group N―C=O as exists in many KHIs,and the lone electron pair of O atom attracts H atom in water molecule to form hydrate bond.Once PEtO binds to the surface of hydrate,O atom in N―C=O group is likely to combine with H atom of water molecule and forms hydrogen bond, which disrupts the previous hydrogen bonds and destroys the hydrate crystal cages.
As can be seen clearly in Fig.3-Fig.7,PEtO polymer absorbs onto the surface of hydrate crystal,the double-bonded O atoms have formed hydrogen bonds with more than one hydrate hydrogen at the same time.On the other hand,there are two side chain ethyls in PEtO,which make the active centers of hydrate crystal segregated and result in steric hindrance to prevent water molecules from contacting with water molecules and makes the cage hard to grow.The active group(N―C=O)adsorbs onto crystal surface forming hydrogen bonds with water molecule.It is likely to display good activity of inhibition.The interactions between hydrate and inhibitor destroy the former perfect neat cages.
4 Conclusions
The decomposing effect of poly(2-ethyl-2-oxazoline)on CH4hydrate was studied by molecular dynamics simulations. The simulation results(RDFs and MSD of O atoms,diffusion coefficient of water molecule)show that PEtO is a good low dosage hydrate inhibitor,which is as active as PVP even at 1%. Within a certain concentration,higher concentration gives rise to better inhibition effect.The polymer binds onto the surface of the CH4hydrate crystal,and starts to interact with water molecules at the interface.The double-bonded oxygen of the active functional group N―C=O combines with H atoms in hydrate and forms hydrogen bonds,resulting in distortion or imperfectness of hydrate cages and the release of trapped CH4molecules.PEtO has two side chain ethyls,which produce steric hindrance.Steric hindrance prevents the decomposing water molecules from regenerating hydrogen bonds with each other. The hydrogen bond displacement by N―C=O group and hydrate cage distortion promote the decomposition of hydrate and the steric hindrance of PEtO makes hydrate hard to grow.
(1) Sum,A.K.;Koh,C.A.;Sloan,E.D.Industrial&Engineering Chemistry Research 2009,48,7457.doi:10.1021/ie900679m
(2) Long,J.P.;Sloan,E.D.Int.J.Thermophys.1996,17,1.doi: 10.1007/BF01448204
(3) Hammerschmidt,E.G.Industrial&Engineering Chemistry 1934,26,851.
(4) Rodger,P.M.;Forester,T.R.;Smith,W.Fluid Phase Equilib. 1996,116,326.doi:10.1016/0378-3812(95)02903-6
(5) Carver,T.J.;Drew,M.G.B.;Rodger,P.M.Phys.Chem.Chem. Phys.1999,1,1807.
(6) Storr,M.T.;Taylor,P.C.;Monfort,J.P.;Rodger,P.M.J.Am. Chem.Soc.2004,126,1569.doi:10.1021/ja035243g
(7) Hawtin,R.W.;Rodger,P.M.J.Mater.Chem.2006,16,1934. doi:10.1039/b600285b
(8) Moon,C.;Hawtin,R.W.;Rodger,P.M.Faraday Discuss.2007, 136,367.doi:10.1039/b618194p
(9) Zhang,J.;Hawtin,R.W.;Yang,Y.;Nakagava,E.;Rivero,M.; Choi,S.K.;Rodger,P.M.J.Phys.Chem.B 2008,112,10608. doi:10.1021/jp076904p
(10) Kvamme,B.;Huseby,G.;Forrisdahl,O.K.Mol.Phys.1997, 90,979.
(11) Kvamme,B.;Kuznetsova,T.;Aasoldsen,K.J.Mol.Graph. Model.2005,23,13.
(12) Kuznetsova,T.;Sapronova,A.;Kvamme,B.;Johannsen,K.; Haug,J.Macromol.Symp.2010,287,168.doi:10.1002/ masy.201050124
(13) Wan,L.H.;Yan,K.F.;Li,X.S.;Fan,S.S.Acta Phys.-Chim. Sin.2009,25,486.[万丽华,颜克凤,李小森,樊栓狮.物理化学学报,2009,25,486.]doi:10.3866/PKU.WHXB20090315
(14) Balbuena,D.A.G.;Balbuena,P.B.J.Phys.Chem.C 2007,111, 15554.doi:10.1021/jp071959c
(15) Anderson,B.J.;Radhakrishnan,R.;Tester,J.W.;Trout,B.L. Abstr.Am.Chem.Soc.2005,229,U593.
(16) Zhang,M.;Anderson,B.J.;Warzinski,R.P.;Holder,G.D. Molecular Dynamics Simulation of Hydrate Lattice Distortion. Prepr.Pap.Am.Chem.Soc.,Div.Fuel Chem.,Salt Lake City, 2009;p 237.
(17) Colle,K.S.;Oelfke,R.H.;Kelland,M.A.Polymer contg. amide unit|used for inhibiting formation of gas hydrate(s)in e.g. oil or gas pipeline;GB Patent 2301824-A,1996-12-18.
(18) Karaaslan,U.;Parlaktuna,M.Energy&Fuels 2002,16,1387. doi:10.1021/ef0200222
(19) Geng,C.Y.;Wen,H.;Zhou,H.J.Phys.Chem.A 2009,113, 5463.doi:10.1021/jp811474m
(20) Kirchner,M.T.;Boese,R.;Billups,W.E.;Norman,L.R.J.Am. Chem.Soc.2004,126,9407.doi:10.1021/ja049247c
(21) McMullan,R.K.;Jeffrey,G.A.J.Chem.Phys.1965,42,2725. doi:10.1063/1.1703228
(22) Berendsen,H.J.C.;Grigera,J.R.;Straatsma,T.P.J.Phys. Chem.1987,91,6269.doi:10.1021/j100308a038
(23) Materials Studio,Version 4.4;Accelrys Software Inc:San Diego,2008.
(24) Tse,J.S.;Klein,M.L.;McDonald,I.R.J.Phys.Chem.1983, 87,4198.doi:10.1021/j100244a044
(25) Moon,C.;Taylor,P.C.;Rodger,P.M.J.Am.Chem.Soc.2003, 125,4706.doi:10.1021/ja028537v
(26) Kelland,M.A.J.Appl.Polym.Sci.2011,121,2282.doi: 10.1002/app.33942
(27)Ajiro,H.;Takemoto,Y.;Akashi,M.;Chua,P.C.;Kelland,M.A. Energy&Fuels 2010,24,6400.doi:10.1021/ef101107r
(28) Chen,Y.J.;Wang,Y.H.;Fan,S.S.;Lang,X.M.Acta Chimica Sinica 2010,68,2253. [陈玉娟,王燕鸿,樊栓狮,郎雪梅.化学学报,2010,68,2253.]
December 7,2011;Revised:April 10,2012;Published on Web:April 10,2012.
Molecular Dynamics Simulation of CH4Hydrate Decomposition in the Presence of Poly(2-ethyl-2-oxazoline)
WANG Yan-Hong CHEN Yu-Juan BAO Ling LANG Xue-Mei FAN Shuan-Shi*
(Key Laboratory of Enhanced Heat Transfer and Energy Conservation of the Ministry of Education,School of Chemistry and Chemical Engineering,South China University of Technology,Guangzhou 510640,P.R.China)
Molecular dynamics simulations were carried out to study the decomposition of CH4hydrate in the presence of poly(2-ethyl-2-oxazoline)(PEtO)at different concentrations,including 1.25%,2.50%,and 6.06%(w,mass fraction).The simulation system was composed of a CH4hydrate crystal and PEtO,which contained a 2×2×2 supercell of CH4hydrate crystal and PEtO polymer.System configurations showed that hydrogen bonding networks between water molecules making up the main framework of the hydrate cages were distorted in the presence of the PEtO polymer.Final configurations in all of the systems were completely collapsed.Radial distribution functions of the oxygen atoms,mean square displacements,and diffusion coefficients of water molecules were applied to compare the effect of different PEtO concentrations on the CH4hydrate.Within a certain concentration range,higher concentrations led to a better inhibition effect.It was confirmed that PEtO is a type of prospective low dosage inhibitor with biodegradability.The decomposition mechanism involves the absorption of the PEtO polymer onto the surface of the hydrate crystal,with its active functional group(N―C=O)forming hydrogen bonds with water molecules in the hydrate and decomposing the hydrate surface.PEtO continued to decompose the surface layer of hydrate,resulting ultimately in the collapse of the hydrate cages.
CH4hydrate;Poly(2-ethyl-2-oxazoline);Molecular dynamics simulation; Hydrate inhibitor
10.3866/PKU.WHXB201204113
∗Corresponding author.Email:ssfan@scut.edu.cn;Tel:+86-20-22236581.
The project was supported by the National Natural Science Foundation of China(51106054),Colleges and Universities High-level Talents Program of Guangdong Province,China,and National Key Basic Research Program of China(973)(2009CB219504-03).
国家自然科学基金(51106054),广东省高层次人才项目和国家重点基础研究发展计划(973)(2009CB219504-03)资助ⒸEditorial office ofActa Physico⁃Chimica Sinica
O641
猜你喜欢
西南石油大学学报(自然科学版)(2021年3期)2021-07-16
军民两用技术与产品(2021年10期)2021-03-16
水上消防(2020年1期)2020-07-24
西南石油大学学报(自然科学版)(2018年6期)2018-12-26
疯狂英语·新读写(2018年3期)2018-11-29
纤维复合材料(2018年4期)2018-04-28
中国资源综合利用(2017年4期)2018-01-22
河北地质(2017年2期)2017-08-16
中国塑料(2015年9期)2015-10-14
中国塑料(2014年1期)2014-10-17
