
高含硫污水氯化物测定方法的改进和优化
2012-12-06 00:41郝新焕崔轲龙马红杰
石油化工腐蚀与防护 2012年3期
郝新焕,崔轲龙,马红杰,徐 奕
(中国石油新疆独山子石化分公司,新疆 独山子 833600)
在炼化工业中,原油中的无机氯和有机氯经过水解或分解作用,在一次和二次加工装置的低温部位遇水形成盐酸复合腐蚀环境,促进金属腐蚀,造成碳钢的全面腐蚀、不锈钢材质的孔蚀和应力腐蚀开裂等,严重影响着设备的安全运行,因此需要对设备介质中的氯化物进行分析、监测。
但是在实际监测过程中,对于催化裂化、延迟焦化和加氢裂化等炼油装置的冷凝水样,由于水样性质的复杂性,里面含有大量的硫化物、氨和氮,特别是加氢裂化和炼油的几套加氢装置冷凝水中的硫化物质量浓度高达几千甚至几十万mg/L,对氯化物的测定造成严重的干扰,甚至造成无法检测的情况发生。目前所用的测定水中氯化物的方法是《GB 11896-89氯化物的测定 硝酸银滴定法》,资料和实际情况均显示硫化物、氨氮对氯化物的测定有严重干扰,可通过用过氧化氢处理予以消除,但简单地按照目前方法中硫化物含量高的前处理方法进行处理并不能够解决无法检测的实际问题,因此需要重新寻找新的样品前处理方法,来解决样品中的高硫化物、氨氮含量对氯化物测定的影响。
1 方法的选择
1.1 前处理方法的选择
根据目前的实际情况:氯化物无法检测的样品中硫化物、氨氮含量较高,硫化物质量浓度经常在一千mg/L到几十万mg/L之间,氨氮质量浓度在几百到几十万mg/L之间。大量的硫化物、氨氮的存在对氯化物的测定造成干扰,因此在样品前处理方面,主要针对消除硫化物、氨氮干扰方面对前处理方法进行了筛选,主要筛选出以下四种方法。
1.1.1 前处理 1[1]288
对于水样中含有硫化物、亚硫酸盐或硫代硫酸盐的情况,则加入氢氧化钠溶液将水样调至中性或弱碱性,加入1 mL,30%过氧化氢,摇匀,1 min后加热至70~80℃,以除去过量的过氧化氢。
1.1.2 前处理 2[1]293-294
100 mL水样中加入1+1硫酸,使溶液呈酸性,煮沸5 min除去挥发物,必要时,再加入适量硫酸使溶液保持酸性,然后加入3 mL过氧化氢煮沸15 min,并经常添加蒸馏水从而保持溶液体积在50 mL以上。加入氢氧化钠溶液使呈碱性,再煮沸5 min,冷却后过滤,用水洗沉淀和滤纸过滤,洗涤液和滤液定容后供测定用。亦可在煮沸冷却后定容,静置使沉淀,取上清液进行测定。
1.1.3 前处理 3[2]
取一定体积的水样于250 mL烧杯中,用1mol/L氢氧化钠溶液调节水样至碱性(pH值 =8~9),若水样本身呈碱性则不必加氢氧化钠溶液,置电炉上加热除去NH3,直到蒸汽不再使湿润的pH值试纸变蓝色。再用10%硝酸调节水样至pH值为2 ~6,继续加热除去S2-,直到蒸汽不使湿润的乙酸铅试纸变黑。加入足量的30%过氧化氢除去SO32-,S2O3
2-以及剩余的硫化物等干扰物质,加热过程中适时补充蒸馏水以防烧干。1.1.4 前处理 4[3]
用中速滤纸将样品过滤后,移取滤液10~100 mL于250 mL的三角烧瓶中,加入蒸馏水使总体积约为100 mL和两粒玻璃珠;将三角烧瓶置于电炉上加热,沸腾后微沸7 min以上(溶液变成乳白色或无色);用1 mol/L HNO3滴加溶液至pH值为2~3(用玻璃棒拈此溶液于pH值试纸上观察),微沸2~3 min,接着加入2 mL,30%过氧化氢,继续微沸2~3 min;加1 mol/L NaOH溶液,使溶液呈碱性(用玻璃棒拈此溶液于pH值试纸上观察pH值不小于9);继续微沸5~10 min取下,此时溶液无色透明,(中途若水蒸发过多,可补加一点蒸馏水);水冷至室温后,往溶液中添蒸馏水至50 mL。
1.2 测定方法
氯化物是污水中常见的一种无机阴离子,因适用范围与目的不同测定的方法也有多种,如:容量法、电量法、比色法、比浊法、电位滴定法、电极流动法、红外光谱法和离子色谱法等,其中以容量法、电位滴定法和离子色谱法较为常见,容量法所需设备最为简单,最适合大批量样品的检测,因此该试验采用的方法就是硝酸银滴定法(GB 11896-89)[4]。
在中性至弱碱性范围内(pH值为6.5~10.5),以铬酸钾为指示剂,用硝酸银滴定氯化物时,由于氯化银的溶解度小于铬酸银的溶解度,氯化物首先被完全沉淀出来,然后铬酸盐以铬酸银的形式被沉淀,产生砖红色,达到滴定终点。
溴化物、碘化物和氰化物能与氯化物一起被滴定,正磷酸盐及聚磷酸盐分别超过250 mg/L及25 mg/L时有干扰,铁含量超过10 mg/L时终点不明显。硫化物、硫代硫酸盐和亚硫酸盐对测定造成干扰。
和硝酸汞滴定法在许多方面类似,可以任意选用。适用于较清洁的水样,适用的范围为10~500 mg/L,低于10 mg/L的样品,滴定终点不易掌握,建议采用硝酸汞滴定法。
2 试验部分
2.1 硝酸银滴定法测定
试验采用硝酸银滴定法,前处理分别采用前1~前4进行测定。
2.1.1 仪器设备
(1)(5±0.05)mL酸式滴定管,0.01 mol/L硝酸银标准滴定溶液。
(2)实际样品来源:炼油厂催化裂化、加氢裂化、延迟焦化和加氢联合等装置的高含硫污水。
2.1.2 试验数据
具体试验数据见表1。

表1 试验数据Table 1 Test data mg/L
2.1.3 实验分析
根据试验实际情况结合资料发现:在硝酸银滴定法中:
(1)由于向样品溶液中加入的过氧化氢是过量的,会导致样品溶液煮沸后有未完全溢出的过氧化氢残留,过氧化氢会与铬酸钾反应。做实验,将30%的过氧化氢与铬酸钾指示剂混合,会看到两者发生剧烈的反应,溶液变为棕黑色,同时有大量的气体生成。当样品溶液中有过氧化氢残留时,会出现这样两种情况:残留量较多时,加入铬酸钾指示剂后溶液变为棕黄色,使得滴定无法进行;当溶液中残留较少时,过氧化氢会消耗溶液中的指示剂,从而影响滴定结果。
(2)样品经过氧化氢氧化分解的反应产物中有大量的二氧化碳和氨。二氧化碳和氨在样品溶液煮沸过程中是可以渐渐释放到空气中去的,但当煮沸的时间不够的情况下,试样溶液中就会有这两种物质的残留,且残留量会随煮沸时间的不同而不同,这两种物质会使滴定终点滞后。未溢出的二氧化碳在溶液中以碳酸根形式存在,碳酸根可与银离子生成碳酸银沉淀,氨可与银离子生成银氨络离子,影响滴定结果。
从测定数据来看,采用硝酸银滴定法测定,采用前处理4方法得到的测定结果较好,采用前处理1、前处理2、前处理3时平行测定结果不平行,以至出现滴定终点无法判断和无滴定终点的情况,因此可以看出采用前处理1、前处理2、前处理3不合适。
采用前处理4方法,在实际测定的时候发现:调节溶液的pH值时要精细稳定,要一滴一滴的调,滴定时注意溶液的pH值,最好控制在7.0左右。
2.2 相对标准偏差的测定
采用前处理4+硝酸银滴定法进行多次平行测定,具体数据见表2。
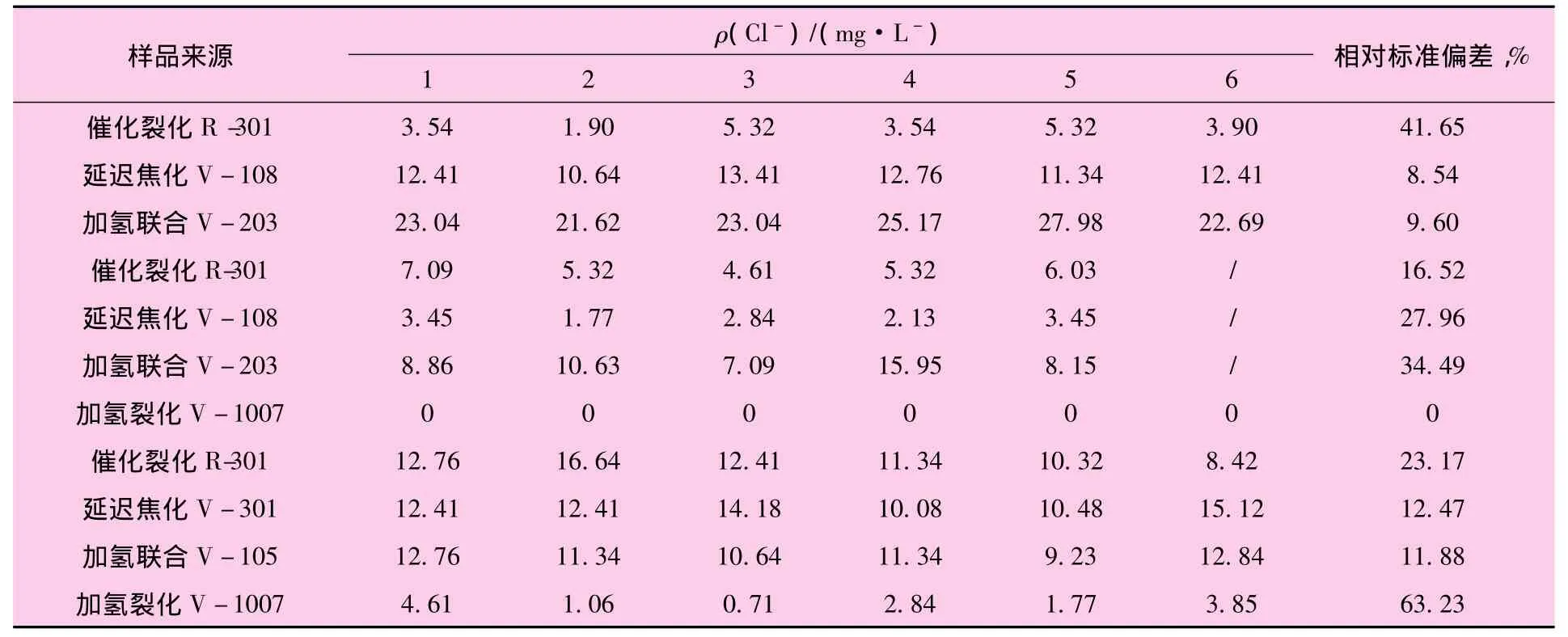
表2 前处理4+硝酸银滴定法相对标准偏差的测定数据Table 2 Pre-treatment+silver nitrate titration method relative standard deviation of themeasured data
采用前处理4+硝酸银滴定法可以检出高硫中的氯化物,但情况不够稳定,相对标准偏差在0%~63.23%之间,考虑到样品处理过程中样品稀释后氯化物含量降低,而硝酸银滴定法的适用范围为10~500 mg/L,而硝酸汞滴定法的适用范围为2.5~500 mg/L,低于10 mg/L的样品,滴定终点不易掌握,因此后面选择采用前处理4+硝酸汞滴定法进行测定。
2.3 采用前处理4+硝酸汞滴定法测定
2.3.1 硝酸汞滴定法[1]290-291
酸化了的样品(pH值为3.0~3.5)以硝酸汞进行滴定时,与氯化物生成难离解的氯化汞。滴定终点时,过量的汞离子与二苯卡巴腙生成蓝紫色的二苯卡巴腙的汞络合物指示终点。硫化物有干扰。
取50 mL水样或经过预处理的水样置于锥形瓶中,另取一锥形瓶加入50 mL蒸馏水作空白试验。加5~10滴混合指示液(结晶二苯卡巴腙和溴酚蓝),摇匀。若试样呈蓝色或红色,则滴加3%硝酸溶液直到溶液转变为黄色后,再多加1 mL。若试样加指示液后立即出现黄色,则滴加1%氢氧化钠溶液至溶液变为蓝色后,逐滴加入硝酸溶液,直到溶液转变为黄色后,再多加1 mL。用0.025 mol/L硝酸汞标准溶液滴定至蓝紫色即为终点。
2.3.2 实际样品的测定
具体数据见表3。
2.3.3 相对标准偏差的测定
进行多次平行测定,具体数据见表4。

表3 前处理4+硝酸汞滴定法测定数据Table 3 Pre-treatment4+mercuric nitrate titration data
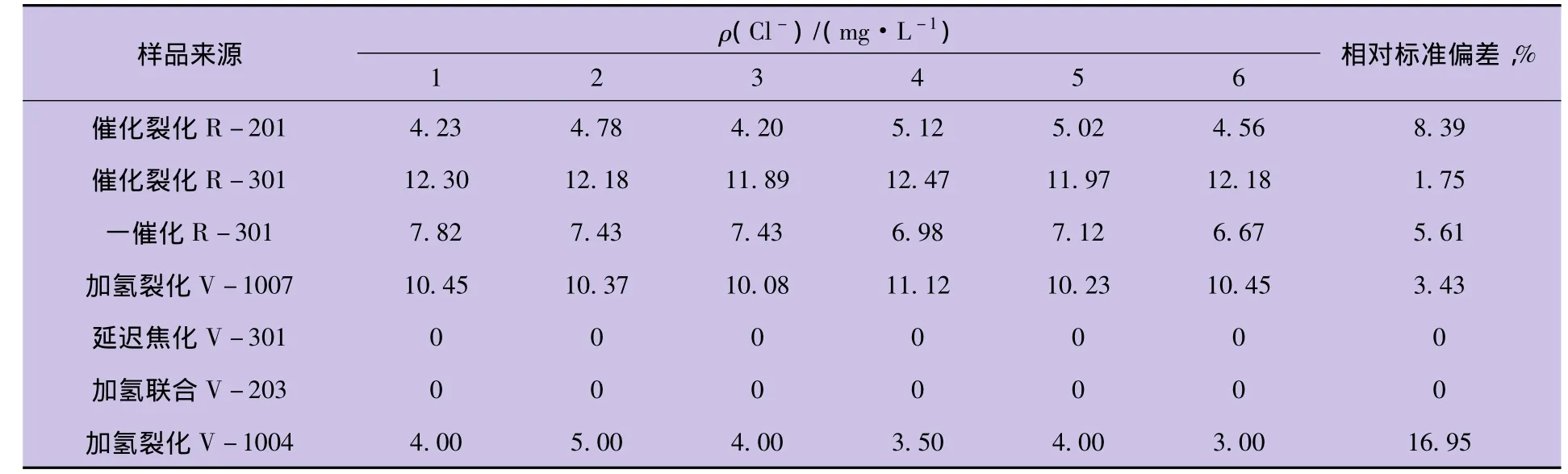
表4 前4+硝酸汞滴定法相对标准偏差的测定数据Table 4 The relative standard deviation of measured data in the first 4+mercuric nitratetitration
2.3.4 加标回收的测定
加标回收率的测定是实验室内经常用以自控的一种质量控制技术,是判定分析结果准确度的量化指标。在理化分析中,用测定加标回收率来反映测试结果的准确度。样品加标回收是指将相同的样品取两份,其中一份加入定量的待测成分标准物质;两份同时按相同的分析步骤分析,加标的一份所得的结果减去未加标一份所得的结果,其差值同加入标准物质的理论值之比即为样品加标回收率。即回收率=加标试样测定值 -试样测定值/加标量×100%。
根据加标回收的要求,加标量不能过大,一般为试样含量的0.5~2倍,加标后的总含量不应超过测定上限,加标物的含量宜较高,加标物的体积应很小,一般以不超过原始试样体积的1%为好,以简化计算,因此结合的检测实际情况选择了100 mg/L的氯化物标液,加入量分别为0.1 mL,0.2 mL,0.5 mL和1.0 mL进行测定。具体数据见表5。

表5 加标回收率的测定数据Table 5 Spiked recoveries of measured data
根据多次试验情况来看,采用前处理4加硝酸汞测定法测定高含硫污水中的氯化物较好,多次平行测定的相对标准偏差在0% ~16.95%,加标回收率在93.85% ~104.59%,结果较好。
3 新方法的建立
综合考虑来看,采用前处理4加硝酸汞滴定法测定高含硫污水中的氯化物较好,所需仪器设备简单,过程容易掌握,适合大批量样品的检测。
总结前面测定的经验,改进后的方法如下:
用中速滤纸将样品过滤后,移取滤液10~100 mL于250 mL的三角烧瓶中,加入蒸馏水使总体积约100 mL和两粒玻璃珠;将三角烧瓶置于电炉上加热,沸腾后微沸7 min以上(溶液变成乳白色或无色);用1mol/L HNO3滴加溶液至pH值为2~3(用玻璃棒拈此溶液于pH值试纸上观察),微沸2~3 min,接着加入2 mL 30%过氧化氢,继续微沸2~3 min;然后用1mol/L NaOH溶液调节,使溶液呈碱性(用玻璃棒拈此溶液于pH试纸上观察pH值不小于9);继续微沸5~10 min取下,此时溶液无色透明,(中途若水蒸发过多,可补加一点蒸馏水);水冷至室温后,往溶液中添蒸馏水至约 50 mL。[2]
取经过预处理的约50 mL水样置于锥形瓶中,加5~10滴混合指示液(结晶二苯卡巴腙和溴酚蓝),摇匀。试样呈蓝色或红色,则滴加3%硝酸溶液直到溶液转变为黄色后,再多加5滴 。若试样加指示液后立即出现黄色,则滴加1%氢氧化钠溶液至溶液变为蓝色后,逐滴加入硝酸溶液,直到溶液转变为黄色后,仔细调节pH值为3.0~3.5。用0.025 mol/L硝酸汞标准溶液滴定至蓝紫色即为终点,同时做空白试验。若氯化物质量浓度小于2.5 mg/L,则改用0.0141 mol/L硝酸汞标准溶液滴定,并使用容量为1 mL的微量滴定管进行。若氯化物质量浓度小于0.1 mg/L,则取适量水样浓缩至大于2.5 mg/L后滴定。
4 实际样品的检测
实际样品检测数据见表6。
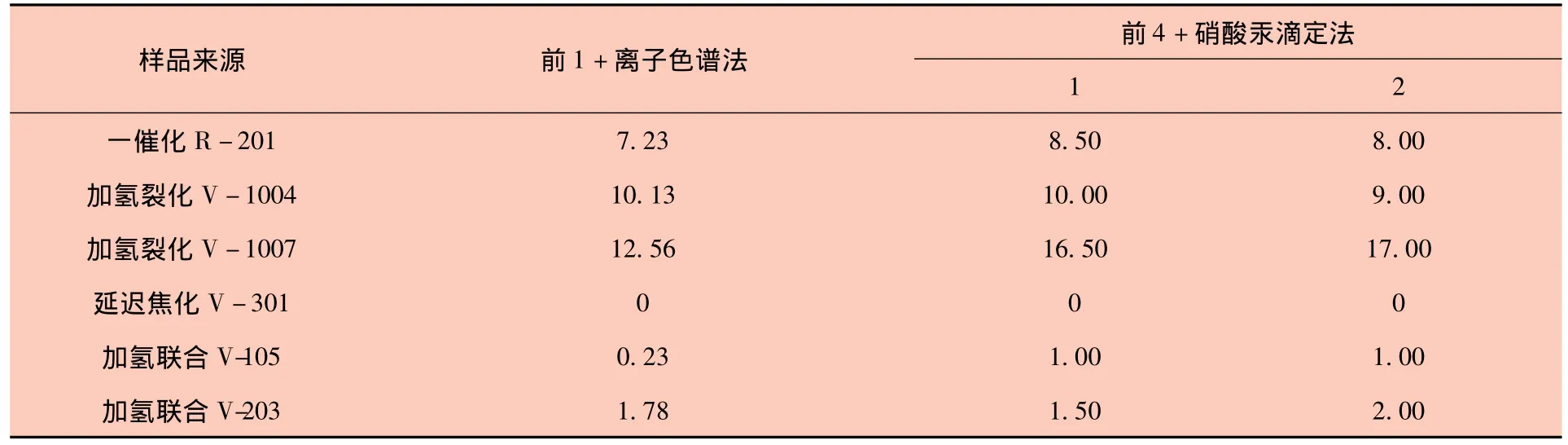
表6 实际样品的测定数据Table 6 The sample analysis data mg/L
5 结论
采用优化后的分析方法(前4+硝酸汞滴定法),通过大量的实际样品的检测和离子色谱法的对照分析,可以看出,优化后的分析方法能很好的检出炼油装置的催化裂化、延迟焦化、加氢裂化等装置的冷凝水样中的氯化物含量,解决了样品中的高硫化物、氨氮含量对氯化物测定的干扰,多次平行测定的相对标准偏差在0% ~16.95%,加标回收率为93.85%~104.59%。
[1]国家环保局《水和废水监测分析方法》编委会.水和废水监测分析方法[M].北京:中国环境科学出版社,1997:288,293-294,290-291.
[2]肖学喜.自动电位滴定法测定炼油工业污水中的氯化物[J].化学分析计量,2007 ,16(5):26~27.
[3]陆克平.汞量法测定炼厂含硫污水中的氯离子的改进[J].中国石油和化工标准与质量,2008(9):25.
[4]中国标准出版社第二编辑室.中国环境保护标准汇编【M】.北京:中国标准出版社,2001:201.
猜你喜欢
云南化工(2021年11期)2022-01-12
食品安全导刊(2021年20期)2021-11-28
环境卫生工程(2021年5期)2021-11-20
疯狂英语·新悦读(2021年5期)2021-06-08
中学生数理化(高中版.高考理化)(2020年3期)2020-05-30
表面工程与再制造(2019年1期)2019-05-11
船电技术(2016年8期)2016-10-27
河北地质(2016年2期)2016-03-20
物理化学学报(2015年5期)2015-02-28
湿法冶金(2014年3期)2014-04-08
