
局灶性脑缺血大鼠预处理后CHOP mRNA及其蛋白的表达☆
2012-11-23 03:02:16胡跃强唐农董少龙刘尊敬祝美珍胡玉英范立雷
中国神经精神疾病杂志 2012年3期
胡跃强 唐农 董少龙 刘尊敬 祝美珍 胡玉英 范立雷
脑缺血预处理(brain ischemic preconditioning,BIP)是近年发现的重要内源性神经保护机制,可使脑组织对以后较长时间的缺血性损伤产生显著的耐受[1]。最近有研究发现内质网应激(endoplasmic reticulum stress,ERS)在此环节中发挥了关键作用,其中 C/EBP 同源蛋白(C/EBP homology protein,CHOP)表达的增加是ERS的标志之一,它参与了细胞凋亡的过程[2]。本研究意在通过检测局灶性BIP后CHOPmRNA及其蛋白在脑内的表达变化,以进一步研究BIP的神经保护机制。
1 材料与方法
1.1 动物分组与处理 健康雄性SD大鼠180只,体质量(250±50)g,随机将大鼠分为3组:假手术组(SO)、大脑中动脉缺血组(MCAO)、脑缺血预处理组(BIP)。 每组按照再缺血后 12 h、1 d、2 d、3 d四个时间点平均分为4个亚组(n=15)。分别作如下处理:SO组:以假手术代替预缺血及缺血再灌注;MCAO组:以假手术代替预缺血,其余步骤同BIP组;BIP组:MCAO 10 min后抽出栓线,完成预缺血,3 d后再次行MCAO 2 h,再灌注后12 h、1 d、2 d、3 d 处死大鼠。
1.2 动物模型与标本制备 参照Longa的线栓法进行造模。采用大脑中动脉二次线栓法[3]制备大鼠脑缺血预处理模型,结扎左侧颈外动脉远端,在结扎线近端距分又5 mm处剪口,将尼龙线从颈外动脉残端插入至颈总动脉分叉部以上19~20 mm,结扎颈外动脉近心端,10 min后抽出栓线,完成缺血预处理。3 d后再次行MCAO 2 h,再灌注后规定时间点处死动物取脑。
1.3 缺血半暗带脑皮质 CHOP mRNA表达测定 原位杂交法。按试剂盒说明进行操作。采用HMIAS⁃2000高清晰彩色病理图像分析系统测定CHOP mRNA原位杂交染色的灰度值。主要在顶叶缺血半暗带阳性细胞做比较,染色越深,灰度值越小,mRNA表达越多。每张切片测定4个视野(400×),取平均值。
1.4 缺血半暗带脑皮质内CHOP蛋白表达测定 大鼠断头取脑并分离缺血侧顶叶大脑皮质约100 mg,研磨后加入蛋白裂解液,然后4℃12000 r/min 离心 30 min,提取总蛋白,取蛋白样品 40 μg经10%SDS-聚丙烯酰胺凝胶电泳进行分离,然后电转移至PVDF膜上。然后依次加入兔抗CHOP抗体(Bioworld公司),4℃ 孵育过夜;偶联有辣根过氧化物(HRP)羊抗兔IgG(Amersham公司)室温孵育2 h,滴加ECL显色液,Bio⁃Rad凝胶成像仪曝光、显像。采用HMIAS⁃2000高清晰度彩色病理图文分析系统进行图像分析,获取各组Western blot条带的平均光密度(OD)值,内参为GAPDH,目的蛋白与内参光密度比值即为目的蛋白的相对值。
1.5 神经细胞凋亡率检测 大鼠迅速断头取脑,分离缺血半暗带脑组织100 mg,1.25 g/L胰蛋白酶消化后制备单细胞悬液,70%乙醇4℃过夜固定样品,次日离心弃乙醇收集细胞,加入50 mg/L RNase,37℃孵育 30 min,再加入 50 mg/L PI(美国 KPL 公司),4℃避光染色30 min,300目筛网过滤,利用美国贝克曼-库尔特FC 500流式细胞仪计数10000个细胞,测定各期细胞DNA含量并计算凋亡细胞所占比例。
1.6 统计学方法 采用SPSS13.0处理数据。所有数据以表示,组间比较用方差分析进行检验,检验水准α=0.05。
2 结果
2.1 CHOP mRNA表达变化 原位杂交显示:SO组有少量CHOP mRNA表达;MCAO组大鼠脑缺血再灌注后12 h缺血半暗带CHOP mRNA表达达高峰(P<0.01),随再灌注时间延长其表达逐渐下降,但仍保持较高表达水平 (P<0.01);BIP组缺血后各时间点其表达水平较MCAO组均明显下降(P<0.05)。 见表 1、图 1。

表1 大鼠顶叶皮层CHOP mRNA表达的灰度值
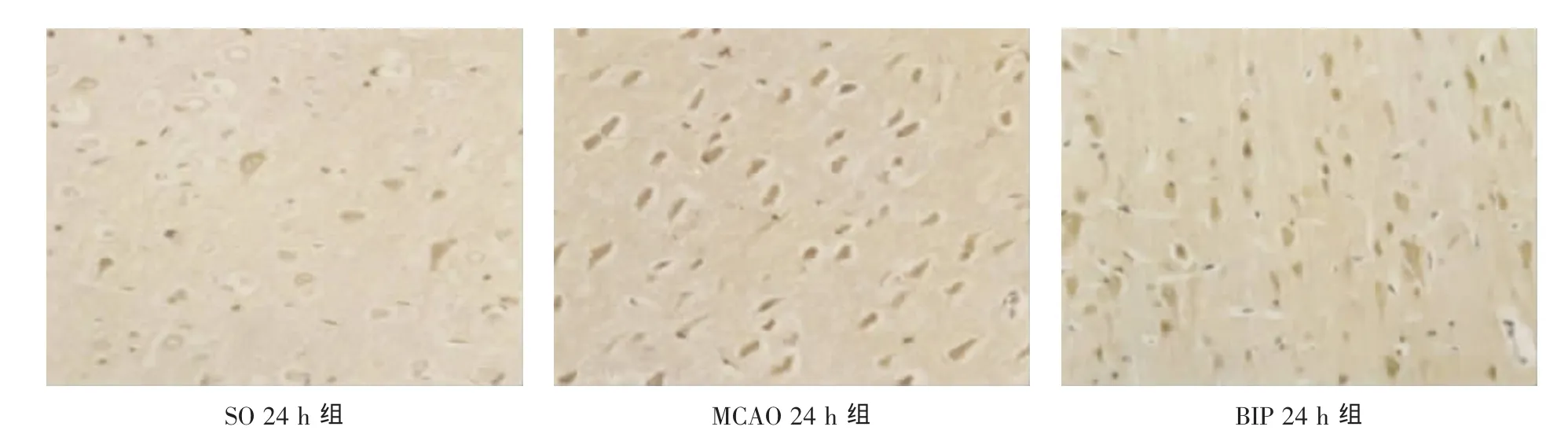
图1 缺血半暗带CHOP mRNA的表达(原位杂交)
2.2 CHOP蛋白表达的变化 Western blot显示:SO组有少量CHOP蛋白表达;MCAO组12 h缺血半暗带CHOP蛋白表达开始显著增加,24 h达高峰(P<0.01),随再灌注时间延长其表达逐渐下降,但仍保持较高表达水平(P<0.01);BIP组缺血各时间点其表达水平均显著降低(P < 0.05,P < 0.01)。见表 2、图 2。

表2 大鼠顶叶皮层CHOP蛋白表达的光密度值

图2 CHOP蛋白的表达(Western blot)
2.3 神经细胞凋亡率变化 流式细胞术细胞周期定量分析表明,SO组未见明显的凋亡峰,其细胞凋亡百分率很低。脑缺血再灌注12 h后,MCAO组缺血半暗带细胞凋亡发生率较SO组显著增加(P<0.01),1 d时达到高峰,以后时间点逐渐下降,但仍高于SO组(P<0.01);BIP组各个时间点神经元凋亡发生率较MCAO组显著降低(P<0.05,P<0.01)。说明BIP具有减轻脑缺血再灌注后神经元凋亡的作用。见表3。
3 讨论
脑缺血预处理(BIP)是指对脑组织采用机械刺激,如一次或多次短暂性脑缺血再灌注后,诱导脑组织产生内源性保护机制,使其对以后较长时间的缺血性损伤产生显著的耐受,这种现象又称为脑缺血耐受。最近有研究发现BIP可激发适当的内质网应激(ERS),增强细胞耐受后继长时间缺血刺激的能力,延缓或减轻I/R造成的组织损伤,并认为ERS可能是脑缺血细胞损伤的关键环节[4]。脑缺血后ERS激活并可诱导CHOP/Gadd153等未折叠蛋白反应 (UPR)靶基因在缺血后表达[5],引起细胞凋亡。其可能机制如下:①内质网跨膜蛋白IRE1和ATF6活化[6],其胞浆活性部分进入核内,与ERS反应元件相连,启动CHOP转录与表达[7];②通过PERK-eIF2a途径活化,导致转录因子ATF4和ATF3表达,前者结合氨基酸调控元件,再诱导CHOP表达。CHOP可能通过下调Bcl-2表达、耗竭谷胱甘肽、促进活性氧族产生等,活化Caspase-12,最终导致细胞凋亡[8]。CHOP基因敲除可增强细胞对抗ERS所致凋亡的能力;相反,CHOP过度表达的细胞则对ERS所致凋亡更为敏感。

表3 各组大鼠神经细胞凋亡率
己有研究在脑缺血再灌动物模型中检测出CHOP表达增加。如Yuan等[9]制作大鼠短暂性全脑缺血模型,阻断脑血流30 min后再灌注,结果显示再灌注6 h至24 h大脑皮层缺血半暗带CHOP蛋白表达逐渐增加,再灌注24 h达高峰;Jin等[10]利用微阵列分析技术发现大鼠海马部位CHOPm⁃RNA 在再灌注 4 ~ 24 h 表达增加;Tajiri等[11]将野生型及CHOP-/-小鼠制成永久性脑缺血模型,检测CHOPmRNA及其蛋白在野生型小鼠纹状体及海马的表达以,实验发现术后12 h CHOPmRNA表达显著增加,术后24 h在受损神经元胞核内检出CHOP蛋白表达,而CHOP-/-小鼠7 d后海马和纹状体的神经细胞死亡较对照组有实质性的减少,这些结果均说明缺血诱导的神经细胞死亡与CHOP有关。
本研究发现,脑缺血120 min再灌12 h缺血侧顶叶皮层CHOPmRNA表达达高峰,随再灌注时间延长其表达逐渐下降,再灌注72 h仍可见其高表达,其蛋白表达于再灌24 h达高峰,提示大鼠缺血再灌注(ischemia/reperfusion,I/R)损伤诱发的ERS可诱导CHOP的表达。与相关研究报道基本一致[9-10]。结合神经细胞凋亡率在I/R后1 d时达到高峰,提示I/R后CHOP表达上调促进了细胞凋亡。而BIP干预后其表达均有明显的下降,提示BIP可能通过影响ERS后CHOP的表达而抑制细胞凋亡,从而保护神经细胞。其机理有待于进一步研究。
[1]Nakka VP,Gusain A,Raghubir R.Endoplasmic reticulum stress plays critical role in brain damage after cerebral ischemi⁃a/reperfusion in rats[J].Neurotox Res,2010,17 (2):189-202.
[2]Liu XQ,Sheng R,Qin ZH.The neuroprotective mechanism of brain ischemic preconditioning[J].Acta Pharmacol Sin,2009,30(8):1071-1080.
[3]郝玉曼,罗祖明,周东.局灶预缺血诱导脑缺血耐受的动物模型[J].中风与神经疾病杂志,2003,20(2):129-130.
[4]DeGracia DJ,Montie HL.Cerebral ischemia and the unfolded protein response[J].J Neurochem,2004,91(1):1-8.
[5]宋小燕,赵永波,周晓琳,等.大鼠脑缺血再灌注后内质网应激相关因子表达的改变[J].中国神经精神疾病杂志,2007,33(10):624-626.
[6]Rissanen A,Sivenius J,Jolkkonen J.Prolonged bihemispheric alterations in unfolded protein response related gene expression after experimental stroke[J].Brain Res,2006,1087(1):60-66.
[7]Oida Y,Shimazawa M,Imaizumi K,et al.Involvement of en⁃doplasmic reticulum stress in the neuronal death induced by transient forebrain ischemia in gerbil[J].Neuroscience,2008,151(1):111-119.
[8]Badiola N,Penas C,Miñano⁃Molina A,et al.Induction of ER stress in response to oxygen⁃glucose deprivation of cortical cul⁃tures involves the activation of the PERK and IRE⁃1 pathways and of caspase⁃12[J].Cell Death Dis,2011,2(4):e149.
[9]Yuan Y,Guo Q,Ye Z,et al.Ischemic postconditioning pro⁃tects brain from ischemia/reperfusion injury by attenuating en⁃doplasmic reticulum stress⁃induced apoptosis through PI3K⁃Akt pathway[J].Brain Res,2011,1367(1):85-93.
[10]Jin K,Mao XO,Eshoo MW,et al.Microarray analysis of hippocampal gene expression in global cerebral ischemia [J].Ann Neurol,2001,50(l):93-103.
[11]Tajiri S,Oyadomari S,Yano S,et al.Ischemia⁃induced neu⁃ronal cell death is mediated by the endoplasmic reticulum stress pathway involving CHOP [J].Cell Death Differ,2004,11(4):403-415.
猜你喜欢
山东医药(2024年2期)2024-03-26 14:28:59
现代临床医学(2022年1期)2022-02-12 02:04:36
中成药(2021年5期)2021-07-21 08:39:04
中国神经精神疾病杂志(2021年6期)2021-03-28 00:42:06
中国医学影像技术(2019年3期)2019-03-25 03:45:52
中国CT和MRI杂志(2016年11期)2017-01-18 10:57:05
安徽农业科学(2015年10期)2015-04-24 08:19:26
新农村(2014年12期)2015-04-08 02:32:48
发明与创新(2015年37期)2015-02-27 10:40:25
癌变·畸变·突变(2014年3期)2014-03-01 04:39:48
