
硬叶兜兰花芽SSH文库的构建
2012-11-14 08:39:09王燕君闻真珍谭志勇王亚平刘运权
华南师范大学学报(自然科学版) 2012年1期
王燕君,闻真珍,张 娥,谭志勇,王亚平,刘运权,刘 伟*
(1.东莞市农业种子研究所,广东东莞 523063; 2.华南农业大学生命科学学院, 广东广州 510642)
硬叶兜兰花芽SSH文库的构建
王燕君1,闻真珍2,张 娥2,谭志勇1,王亚平1,刘运权2,刘 伟2*
(1.东莞市农业种子研究所,广东东莞 523063; 2.华南农业大学生命科学学院, 广东广州 510642)
利用SMART策略构建了硬叶兜兰花芽的抑制性消减杂交(SSH)文库.通过PCR对文库中插入的片段进行检测后,筛选了288个插入片段为500 bp以上的克隆进行测序.测序结果去除载体序列后聚类得到18条差异表达片段,用BLAST进行比对分析表明,这些差异表达基因所编码的蛋白涉及光合作用、合成代谢、基因调控等功能,其中,包括多个转座子和反转录转座子的同源基因.
硬叶兜兰(Paphiopedilummicranthum);抑制消减杂交(SSH);花发育
兜兰(Paphiopedilum)是兰科植物中最具特色的一个类群,唇瓣呈兜状,背萼发达,具有艳丽的花纹.兜兰的两枚能育雄蕊着生在蕊柱的两侧,发达的背萼呈扁圆形或倒心形,在各瓣中显著.在观赏兜兰生产中,种苗生产和栽培技术都不完善,而花期调控问题也难于解决.
目前对兜兰的研究集中在生理生态、栽培和繁殖技术等方面[1-4],对其花发育的分子生物学研究尚为空白.本研究以硬叶兜兰(Paphiopedilummicranthum)为材料,利用抑制性消减杂交技术(suppression subtractive hybridisation, SSH),以花芽mRNA(tester,试验方)及营养芽mRNA(driver,驱动方)为样品,构建硬叶兜兰花芽SSH文库,分离与其花发育相关的差异表达基因片段,并对相关片段的功能进行初步分析,以期为阐明硬叶兜兰花发育的分子机制提供重要的信息.
1 材料和方法
1.1材料
硬叶兜兰栽培于华南农业大学生命科学学院温室.于不同发育时期取其营养芽和花芽,-80℃冰箱中保存以备提取总RNA.
试剂和酶:SMARTTMcDNA Library Construction Kit为Clontech公司产品;焦碳酸二乙酯(DEPC),MOPS,氨卞青霉素钠盐,TaqDNA聚合酶,各种限制性内切酶和T4 DNA Ligase均购自上海Sangon公司;其它试剂为国产分析纯.
1.2方法
1.2.1 总RNA提取 总RNA采用Trizol法提取,利用0.8%的琼脂糖凝胶电泳进行检测,利用紫外分光光度法检测质量和浓度.
1.2.2 cDNA合成及消减文库的建立 采用Clontech公司的PCR-SelectTMcDNA Subtraction Kit中的试剂及反转录酶,合成双链cDNA.以花芽为Tester,营养芽为Driver.按照试剂盒说明书将上述合成的双链cDNA进行酶切、接头连接和消减杂交,稀释的杂交产物用primer1进行32个循环的第一轮PCR,产物稀释后用巢式PCR引物进行32个循环的第二轮扩增.2轮扩增后得到的PCR产物经过切胶回收纯化后,连接到pMD18-T载体并转化大肠杆菌DH5α,通过蓝白斑筛选阳性克隆并计算转化率.
1.2.3 消减效率的分析 分别以消减和未消减的样品为模板,用上游引物5′-ACCACAGTCCATGCCATCAC-3′和下游引物5′-TCCACCACCCTGTTGCTGTA-3′进行PCR扩增甘油醛-3-磷酸脱氢酶(G3PDH)基因,对18、23、28和33个循环的PCR产物进行琼脂糖凝胶电泳检测,根据G3PDH基因表达量的变化分析所建文库的消减效率.
1.2.4 测序及差异表达基因功能分析 挑取阳性克隆,用M13通用引物检测插入片段.将插入片段大小为500 bp以上的克隆送到上海生工测序.其测序结果去除载体序列后,用DNAstar进行聚类,通过BLASTx比对分析,预测相应基因的功能及其与硬叶兜兰花发育的关系.
2 结果与分析
2.1硬叶兜兰营养芽和花芽总RNA质量检测
电泳检测结果表明,28S和18S rRNA条带清晰,说明提取的总RNA完整(图1).经紫外分光光度检测,得到营养芽总RNA样品的OD260/OD280为1.89,花芽的为1.85,说明RNA纯度较高,可用作后续反转录的模板.
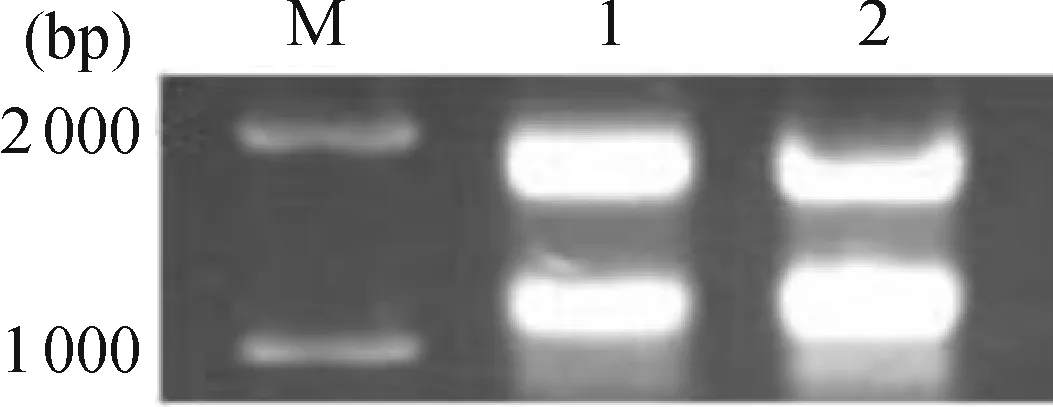
M: DL2000 ladder; 1: total RNA extracted from vegetative buds; 2: total RNA extracted from floral buds.
图1 硬叶兜兰营养芽和花芽总RNA
Figure 1 Total RNA extracted from vegetative and floral buds ofP.micranthum
2.2cDNA合成及消减文库质量检测
合成的cDNA相对分子量大小分布于500~2 250 bp之间,呈均匀弥散状态(图2),质量符合后续酶切实验的要求.酶切后的花芽cDNA片段加上接头,作为tester,未加接头的营养芽cDNA片段为driver,杂交后的cDNA进行2轮PCR扩增.为分析消减效率,分别以消减及未消减PCR 产物为模板,用G3PDH引物进行PCR 扩增,分别对18、23、28和

M: 250bp DNA ladder; 1:double-strand cDNAs of vegetative buds; 2: double-strand cDNAs of vegetative buds
图2 硬叶兜兰营养芽和花芽双链cDNA
Figure 2 Double-strand cDNAs of vegetative and floral buds ofP.micranthum
33个循环的PCR产物进行电泳分析,结果显示:与未消减组PCR产物相比,消减组PCR产物中G3PDH 基因产物大大减少,说明所构建的消减文库有一定的消减效率(图3).

图3 消减效率分析结果
经过2轮PCR后,差异片段得到富集(图4),集中分布于750~1 500bp之间(图4,Lane1).将第2轮PCR产物纯化,转化大肠杆菌DH5α后共获得445个阳性克隆.选取其中320个阳性克隆,以M13通用引物进行PCR,电泳检测出具有1个插入片段的克隆300个,阳性克隆率93.75%;其中插入的片段大小位于250~1 500 bp之间(图5).
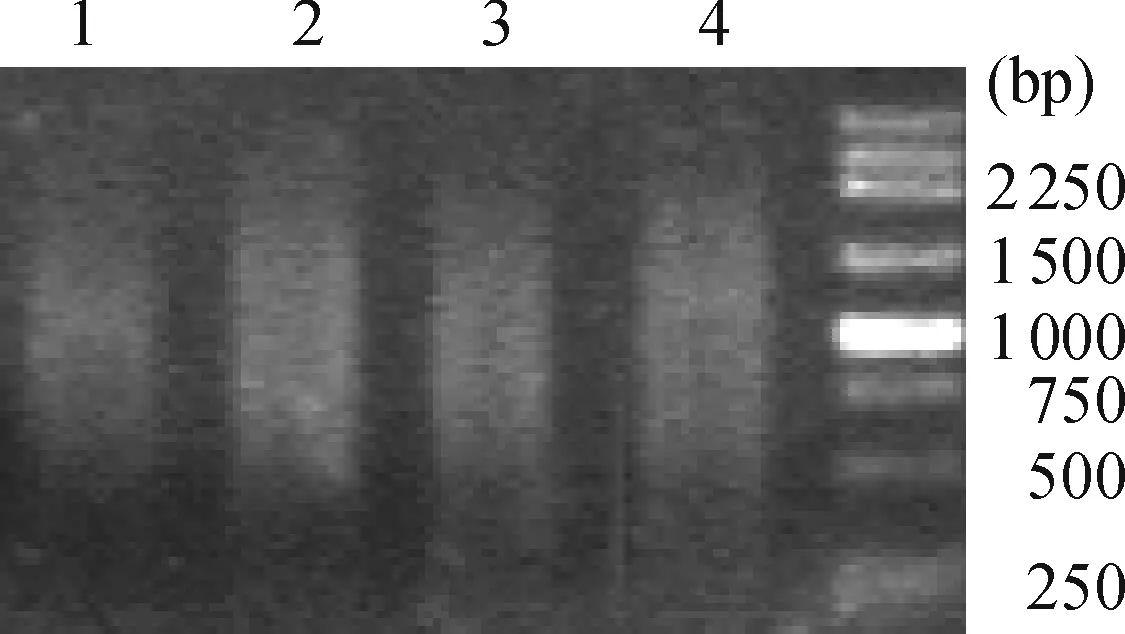
M: DL2000 ladder; 1:secondary PCR product after subtraction; 2: secondary PCR product of un-subtracted samples; 3:primary PCR product after subtraction; 4: primary PCR products of un-subtracted samples
图4 消减PCR电泳检测结果
Figure 4 Electrophoresis analysis of subtracted PCR products
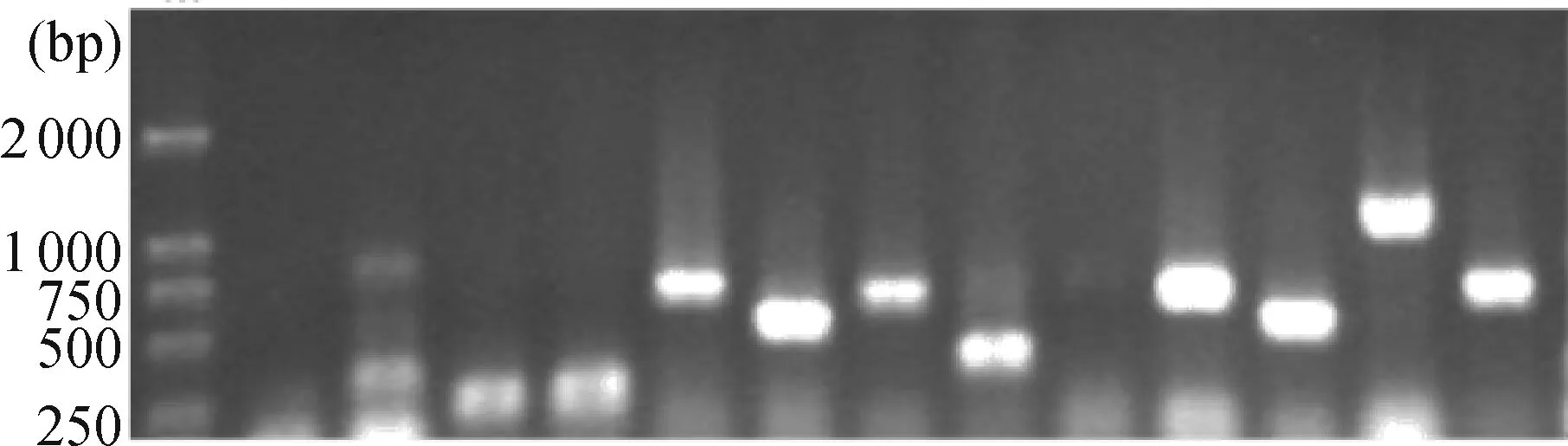
图5 文库中部分阳性克隆PCR鉴定结果
Figure 5 PCR products of some randomly selected subtracted-library clones
2.3EST分析
EST结果见表1.在聚类后的18个EST序列中,3个没有同源匹配基因,5个为未知功能的假定蛋白,其余具有同源序列的特异表达基因包括与植物细胞合成代谢相关的糖原合成酶基因、光合作用相关基因、基因调控信号途径相关激酶基因以及与mRNA修饰相关的基因等.在同源序列比对中,有多个与反转录转座子相关的同源序列,另外还包含1个转座子序列和1个可作为分子标记的微卫星序列.

表1 硬叶兜兰花芽中特异表达的序列Table 1 Sequences expressed differentially in floral buds of P. micranthum
3 讨论
SSH是一种以抑制PCR为基础的cDNA 缩减杂交法,1999年BIRCH等[1]第一次将此技术成功运用于对马铃薯晚疫病的研究中.随后,在兰花特异基因的分离方面,研究者利用SSH技术成功获得了文心兰假鳞茎中的差异表达基因[2]、与万代兰(VandaMimi Palmer)香气相关的基因[3]以及蝴蝶兰唇瓣突变体中的差异表达基因[4],本研究采用SMART cDNA 合成技术,利用没有纯化的硬叶兜兰总RNA,成功的构建了硬叶兜兰花芽的SSH文库,在此基础上分离得到的在花芽中特异表达的基因,将为研究硬叶兜兰花发育的分子机理提供基础.
花发育的分子机理是近二十多年来植物学领域的研究热点,通过对模式植物拟南芥的研究,提出了植物开花的几种途径以及花器官发育的ABC模型[5].然而由于兰花基因组信息的缺失及其转化再生体系的不完善,兰花花发育分子基础的研究落后.研究者从蝴蝶兰、文心兰以及石斛兰中克隆到了大量的与花发育相关的基因[6-8],并在经典的ABC模型的基础上,针对蝴蝶兰提出了兰花花器官发育的分子调控模型,认为B类MADS-box基因在调控兰花花器官的发育过程中起到关键作用[9].花期调控方面,已经从石斛兰、蝴蝶兰等兰花中分离到与调控花期相关的基因[6].然而兜兰作为兰花中具有极高观赏价值的成员之一,由于资源的有限和栽培技术、再生体系的不完善,目前的研究大多集中于栽培和繁殖技术方面,对其花发育的研究没有任何相关报道.但花期调控技术却是栽培过程中亟需克服的难题,以硬叶兜兰为例,人工引种栽培后很少开花,导致人工繁殖和育种都成为难题.本研究通过SSH文库筛选硬叶兜兰花芽中的差异表达基因,可以为研究其花发育机理提供基础.但是由于兜兰乃至兰科植物基因组信息的缺失,以及现有数据库中相关信息的匮乏,人们很难根据文库中筛选得到的基因片段在网上搜索到其全部基因信息.因此,本研究中得到的差异表达片段有多数功能未能进行初步确定,需要进一步的实验研究.
通过同源比对,初步确定功能的差异表达基因包括与植物细胞合成代谢相关的糖原合成酶基因和光合作用相关基因PsaB,另外还包括MAPK级联信号通路中的MAPKKK激酶基因.植物中的MAPK级联信号通路参与胁迫信号以及激素信号的传导[9],MAPKKK激酶基因在硬叶兜兰花芽中的差异表达可能与激素信号的传导相关.通过SSH文库筛选到的差异表达基因包含一个与RNA剪切相关的基因,该基因的表达与转录后的调控相关.该基因在花芽中差异表达在硬叶兜兰花发育过程中的功能有待进一步研究.
本研究分离的在硬叶兜兰花芽中特异表达的基因中,含有多个转座元件(Transposable elements,TEs)同源基因,其中包括转座子、反转录转座子以及转座酶(表1).TE序列是基因组的重要组成部分,在水稻和拟南芥的基因组中,TE分别占29%[10]和10%[11].HSU等利用细菌人工染色体技术(BAC)对蝴蝶兰的基因组序列进行分析表明,有23%的BESs(BAC end sequences)包括重复DNA序列,其中54.7%为反转录转座子[12].拟南芥中7.8%的EST序列与TE具有同源性,这些基因的产物具有调节功能,但不具有结构蛋白的功能[13].TE可以在植物特定发育阶段和特定组织中表达,参与发育调控.在花发育过程中,也存在TE的特异表达.在春化途径关键基因BvFLC的上游存在2个反转录转座子,DNA甲基化水平的降低可能促进这2个反转录转座子的激活,从而影响下游BvFLC的表达,促进植株抽薹[14].本研究从硬叶兜兰花芽中分离到的TE同源基因,可能参与硬叶兜兰的花发育调控过程,其作用将深入研究,有助于进一步认识TE在植物发育中的调控功能.
[1] BIRCH P R J,AVROVA A O,DUNCAN J M,et al. Isolation of potato genes that are induced during an early stage of the hypersensitive response toPhytophthorainfestans[J]. Mol Plant Microbe In,1999,12(4):356-361.
[2] TAN J,WANG H L,YEH K W. Analysis of organ-specific, expressed genes inOncidiumorchid by subtractive expressed sequence tags library[J]. Biotechnol Lett, 2005,27:1517-1528.
[3] CHAN W S,ABDULLAH J O,NAMASIVAYAM P. Isolation, cloning and characterization of fragrance-related transcripts fromVandaMimi Palmer[J].Sci Hortic ulturae, 2011,127:388-397.
[4] CHEN Y H,TSAI Y J,HUANG J Z,et al.Transcription analysis of peloric mutants ofPhalaenopsisorchids derived from tissue culture[J].Cell Res,2005, 15(8):639-657.
[5] IRISH V F.The flowering ofArabidopsisflower development[J]. Planta, 2010,61:1014-1028.
[6] 陈小强,孙宁,刘玉芹,等.兰花花发育的分子生物学研究[J].天津农学院学报,2011, 18(1):27-31.
[7] 王亚琴,钟开新,阳成伟,等.石斛兰SIZ1基因的克隆及序列分析[J].华南师范大学学报:自然科学版,2011(2):119-123.
[8] 梁山,陈莉莉,李洪清.基于金钗石斛EST的短串联重复序列的挖掘[J]. 华南师范大学学报:自然科学版,2011(2):113-118.
[9] 陈娅斐,冯斌,赵小明,等.MAPK级联途径在植物信号传导中的研究进展[J].植物学通报,2005,22(3):357-365.
[10] MESSING J,BHARTI A K,KARKIWSKI W M,et al.Sequence composition and genome organization of maize[J].PNAS,2004,101:14349-14354.
[11] ARABIDOPSIS G I. Analysis of the genome sequence of the flowering plantArabidopsisthaliana[J].Nature,2000,408:796-815.
[12] HSU C C, CHUNG Y L, LEE Y L, et al. An overview of thePhalaenopsisorchid genome by BAC end sequence analysis[J]. BMC Plant Biol, 2011, 11:3.
[13] LOCKTON S, GAUT B S. The contribution of transposable elements to expressed coding sequence inArabidopsisthaliana[J]. J Mol Evol, 2009, 68:80-89.
[14] TRAP-GENTIL M V, HÉBRARD C, LAFON-PLACETTE C, et al. Time course and amplitude of DNA methylation in the shoot apical meristem are critical points for bolting induction in sugar beet and bolting tolerance between genotypes[J]. J Exp Bot, 2011, 62(8):2585-2597.
Keywords:Paphiopedilummicranthum; suppression subtractive hybridization (SSH); floral development
ConstructionofSuppressionSubtractiveHybridizationLibraryfromtheFloralBudsofPaphiopedilumMicranthum
WANG Yanjun1, WEN Zhenzhen2, ZHANG E2, TAN Zhiyong1, WANG Yaping1, LIU Yunquan2, LIU Wei2
(1. Institute of Agricultural Seeds, Dongguan 523063, China; 2. School of Life Science, South China Agricultural University, Guangzhou 510642, China)
With high ornamental and research value,Paphiopedilummicranthumis a species of the genusPaphiopedilumnative to China, which is endangered and protected intensively by the country. However, there are few reports on its floral development mechanism because of the supply shortage and backward cultivation technology. In this study, a suppression subtractive hybridization (SSH) library was constructed using the cDNAs from the floral and vegetative buds ofP.micranthumas the tester and the driver, respectively. The insert fragments were tested by PCR, and 288 clones with 500 bp or longer from the library were selected for sequencing. Totally, 18 ESTs were obtained, after vector sequences removed and clustered. The ESTs were functionally annotated by Blast. Those which matched significantly with Nr database were further classified into functional groups including photosynthesis, biosynthetic metabolism, gene regulation, etc. Among them, a few sequences with homology to transposons or retro-transposons were obtained. The genes differentially expressed in floral buds ofP.micranthumisolated in this study could make a base for investigating the molecular mechanism of floral development.
2011-08-31
广东省科技计划项目(2008B020400006);广东省良种培育和引进专项资金项目(2008-810);东莞市高等院校科研机构科技计划项目(2007108101107)
*通讯作者,liuwei@scau.edu.cn
1000-5463(2012)01-0099-04
Q945.4
A
【责任编辑 成 文】
猜你喜欢
红领巾·萌芽(2024年3期)2024-04-22 04:40:32
红领巾·萌芽(2024年3期)2024-04-22 04:40:32
广西科学院学报(2022年2期)2022-07-14 03:55:52
林业科学(2022年1期)2022-03-23 06:56:24
中国蜂业(2021年5期)2021-05-22 02:59:26
猪业科学(2021年3期)2021-05-21 02:05:36
幽默大师(2020年10期)2020-11-10 09:07:22
中华诗词(2019年1期)2019-11-14 23:33:56
猪业科学(2018年4期)2018-05-19 02:04:31
浙江农林大学学报(2016年6期)2016-12-12 12:01:32
