
甲基异丙基酮制备3,4-二甲基吡唑的热力学计算与分析
2012-11-06 04:33王莎莎郭瓦力刘思乐王修纲
当代化工 2012年1期
王莎莎,郭瓦力,田 旭,冯 健,李 萍,刘思乐,王修纲
(沈阳化工大学,辽宁 沈阳 110142)
甲基异丙基酮制备3,4-二甲基吡唑的热力学计算与分析
王莎莎,郭瓦力,田 旭,冯 健,李 萍,刘思乐,王修纲
(沈阳化工大学,辽宁 沈阳 110142)
采用Joback基团贡献法、Lydersen法、Riedel法、Ednister法、Rowlinson-Bondi对应状态法、Benson法估算了甲基异丙基酮制备二甲基吡唑反应体系相关物质的基本物性参数如常沸点、临界参数和偏心因子,基本热化学性质参数如气体热容、液体热容和蒸发潜热、气体标准摩尔熵、相变熵。利用键能法计算了气体的标准反应热,计算了实际反应条件下的反应热、吉布斯自由能及相关反应的化学平衡常数,并对反应体系的特点进行了分析,估算数据可为理论研究、工程设计与放大及生产提供参考。
热力学分析;基团贡献;盖斯定律;3,4-二甲基吡唑
3,4-二甲基吡唑磷酸盐是一种新型硝化抑制剂,它以其显著的硝化抑制效果、较高的氮素利用率、较小的用量、极小的植物残留量且无激素效应、环境友好等特点引起了人们的关注,被誉为最具发展前景的新型高效硝化抑制剂。
硝化抑制剂是缓控释化肥发挥控释作用的重要添加剂之一。2011年,国家发改委将缓控释肥纳进优先发展的行业,农业部首次将缓控释肥列进主推技术,科技部将缓控释肥列入“十二五”科技支撑计划,三部委对发展缓释控肥的高度重视与举措,必将加速缓控释肥的发展与推广,加速化肥行业转型进级,促进我国农业可持续发展。
本文以甲基异丙基酮为原料制备3,4-二甲基吡唑为切入点,结合该反应体系的特点对部分基础热力学数据进行估算和计算,旨在为进一步研究这类复杂反应体系的化学平衡,研究过程进行的方向和限度奠定基础, 并为过程放大、设计及工业化生产提供有参考价值的必要数据。这里。
1 制备过程的化学反应
以 C5H10O(甲基异丙基酮)、N2H4·H2O 为原料的3,4-二甲基吡唑制备方法分为两个反应阶段,分别为Step A、Step B。Step A阶段为甲基异丙基酮与水合肼缩合反应阶段,Step B阶段为缩合产物在碘的催化作用下环化阶段。各阶段可能发生的化学反应见表1。
2 热力学数据估算
本文的热力学数据估算主要包括基本物性数据的估算、热化学数据的估算和化学反应热力学数据的计算三部分,其估算过程多采用基团贡献法。
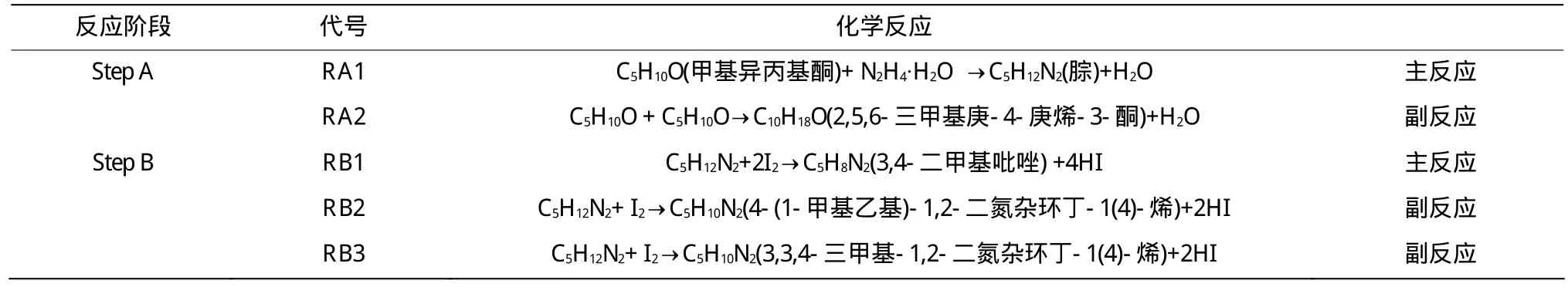
表1 3,4-二甲基吡唑制备过程可能发生的化学反应Table 1 Chemical reactions during preparation process of 3,4-dimethyl-1H-yl-pyrazole
2.1 基本物性数据的估算
本文针对反应体系可能出现的物质进行常沸点、临界参数、偏心因子的估算。
2.1.1 常沸点的估算[1]
常沸点的估算有相对分子量法、有机物估算法、Joback法、修正Joback法、Waston法、和Kinney法,考虑体系中不同物质的结构特点及反应物系多为有机物,本文选用Joback法[1]计算相关物质的常沸点。

2.1.2 纯物质临界参数的估算[1]
纯物质临界参数的估算方法有几十种,较为常用和可靠的是基团法。代表性的基团贡献法有Lydersen法、Ambrose法、Joback法和Fedors法。鉴于Fedors法只适用于临界温度,Ambrose法用于计算支链烃及各种醇时过于繁琐,Joback法包括各种有机物的基团,但未考虑多卤化物间的作用,Lydersen法是推算临界性质最成功有效的基团贡献法之一,本文选用Lydersen法。
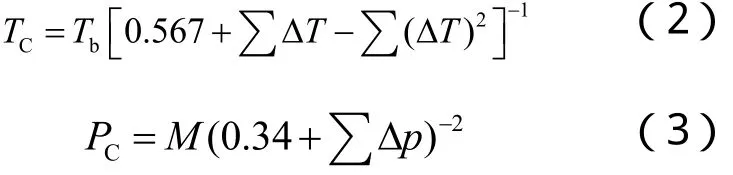
2.1.3 偏心因子的计算[1]
偏心因子的估算可采用Ednister法、Lee-Kesler法和压缩因子法,本文采用Ednister法。

2.1.4 计算举例及数据汇总

甲基异丙基酮及其它物质计算结果见表2。
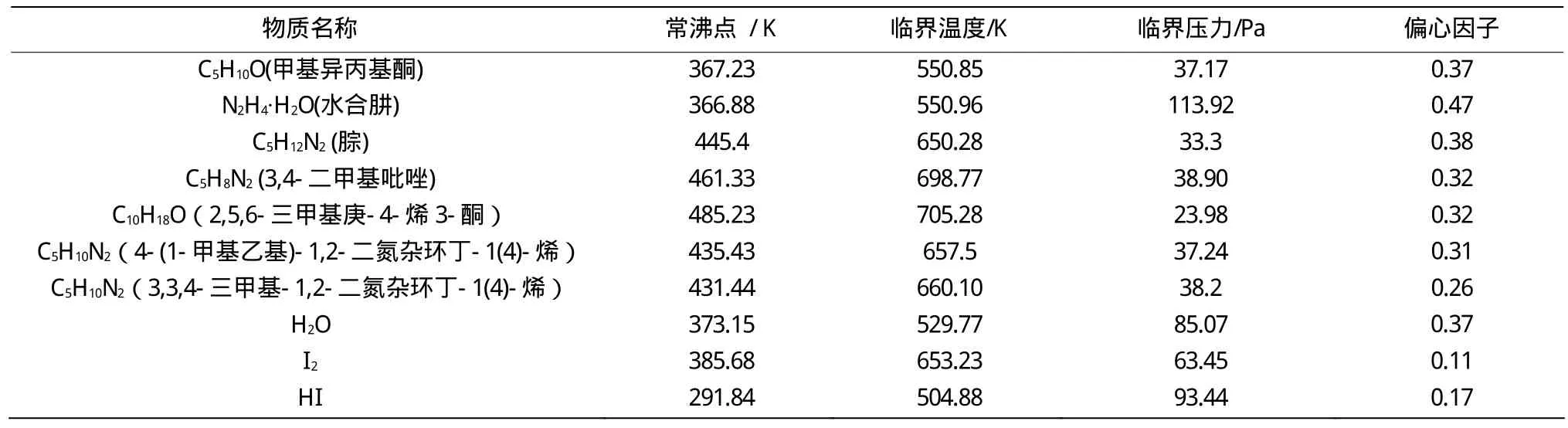
表2 反应体系相关物质的基础物性数据Table 2 Basic property parameters related to the reaction system
2.2 热化学数据的估算
本文估算的相关物质的热化学数据包括:蒸发潜热、气体热容、液体热容、气体标准摩尔熵、相变熵,液体标准摩尔熵等。
2.2.1 蒸发潜热的估算[1]
估算常沸点下的汽化热一般采用Giacalone法、Riedel法、Chen法及Vetere法。本文选择较为简便的Riedel法。
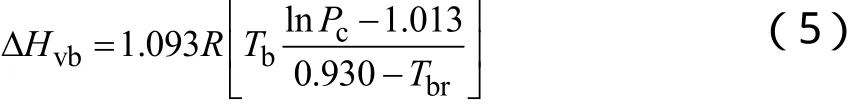
2.2.2 理想气体热容的估算[3]
本文采用Joback基团贡献法进行理想气体热容的估算。
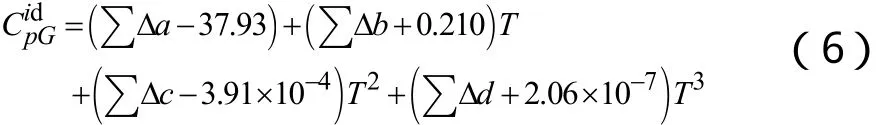
2.2.3 25 ℃液体热容估算
液体热熔估算分为基团贡献法和对应状态法两大类。代表性的基团贡献法有 Chueh-Swanson、Shaw、Missenard法等,代表性的对应状态法包括Rowlinson-Bondi、Sternling-Brown、Yuan-Stiel法等。基团贡献法对某些温度下的热容计算不适用,而对应状态法中的 Rowlinson-Bondi法计算相对简便且能给出不同温度下的热容,所以本文采用Rowlinson-Bondi对应状态法:
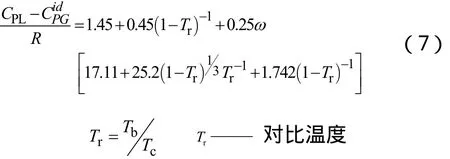
2.2.4 气体标准摩尔熵的计算[3]
气体标准摩尔熵是指标准状态下 1mol纯物质的规定熵。通常采用ABWY法、Thinh法和Benson法估算,本文选择Benson法。计算公式如下:

气体标准摩尔熵计算中,若相关物质无分子对称及光学异构体的,本文以 、h=1计算。
2.2.5 标准摩尔相变熵的计算[4]
本文将常沸点下的相变视为可逆相变,利用盖斯定律设计回路,计算相关物质的标准摩尔相变熵(见图1),为计算25 ℃下液体的标准摩尔熵提供数据。
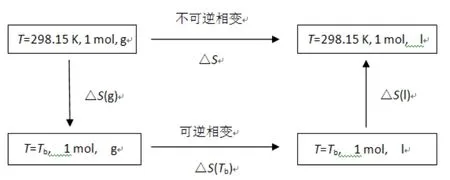
图1 标准摩尔相变熵计算示意图Fig.1 The diagram of standard mole phase transition entropy calculation
由上述的回路设计可得:

2.2.7 计算举例及数据汇总
以甲基异丙基酮(C5H10O)为例计算热力学数据。
(1)汽化热:由(5)式可知
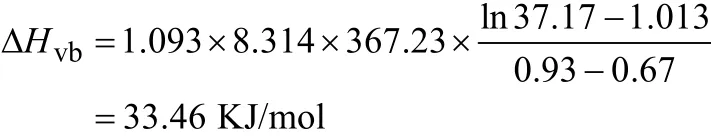
(2)理想气体定压热容:
甲基异丙基酮含有官能团及数据见表3。

表3 甲基异丙基酮含有官能团及数据Table 3 Functional group and data of 3-methyl-2-butanone
所以由(6)式可知:
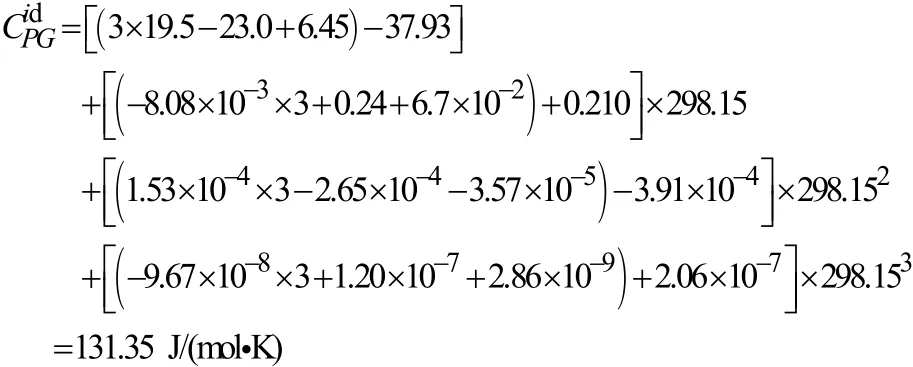
(3) 液体热容:由(7)式可知

(4) 气体标准摩尔熵:
甲基异丙基酮所含基团的DS分别为[2]:127.32、50.53、62.84
所以由(8)式可知:

(5) 标准摩尔相变熵
由甲基异丙基酮的常沸点、蒸发潜热、理想气体热容、液体热容及(10)式可计算其标准摩尔相变熵。
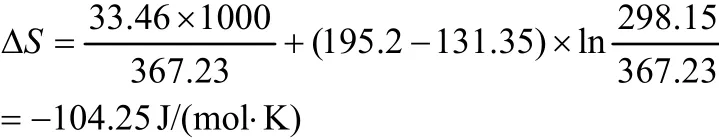
(6) 25 ℃液体的标准摩尔熵

甲基异丙基酮及其它物质的热力学数据见表4。
2.3 化学反应热力学数据的估算
通过化学反应热力学数据的估算可以确定化学反应中能量的转化方式、化学反应的方向性及反应进行的程度。
2.3.1 25 ℃气相反应热的计算[5]
计算示例:以RA1反应为例进行25 ℃气相反应热的计算。
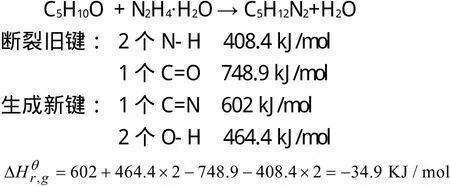
2.3.2 利用盖斯定律进行液相反应热的计算
计算示例:以RA1反应为例进行液相反应热的计算,25 ℃时,液相到气相:
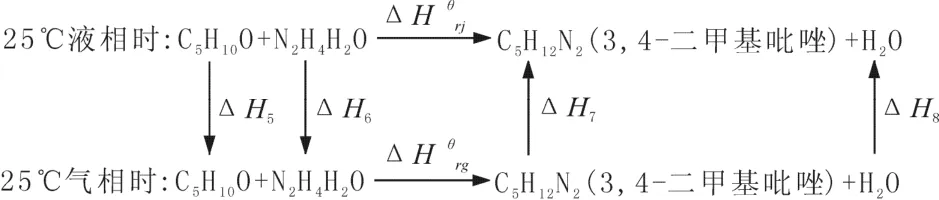
计算所需数据见表4。
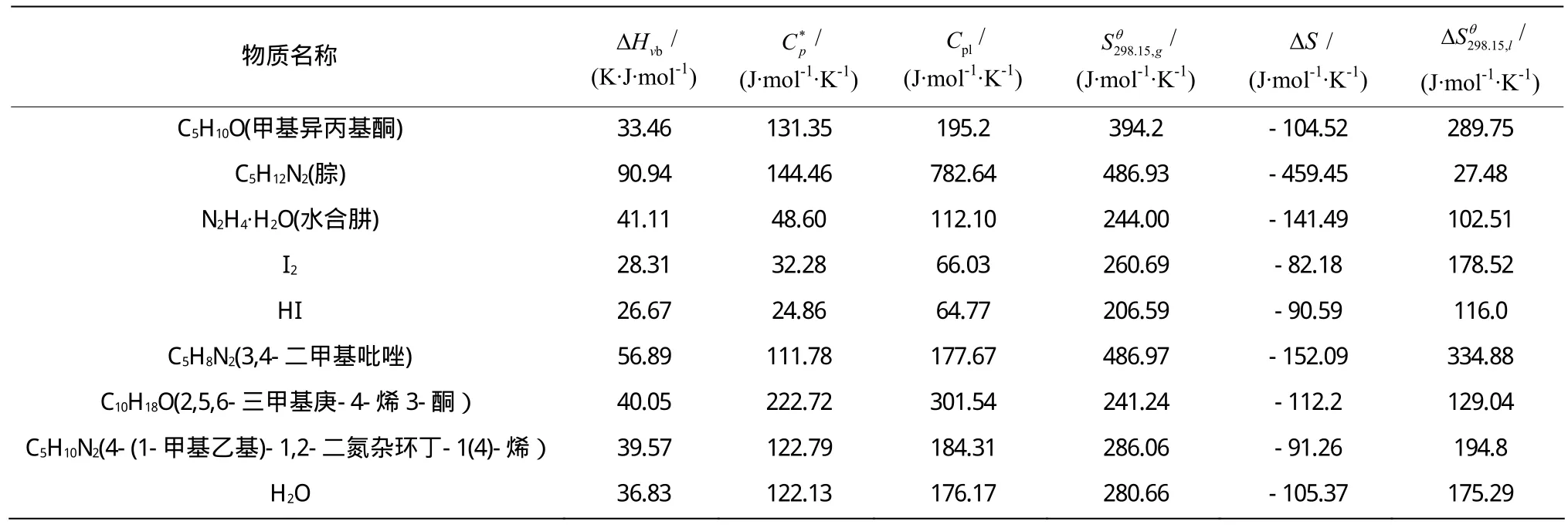
表4 反应体系相关的热化学数据Table 4 Thermal and chemical data related to the reaction systyem
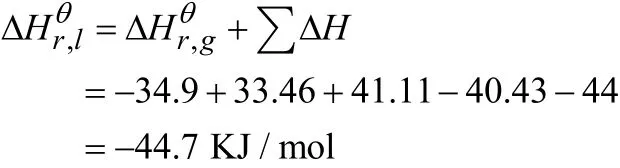
2.3.3 25 ℃下液相吉布斯自由能和平衡常数的计算[2]
吉布斯自由能和平衡常数的计算公式如下:

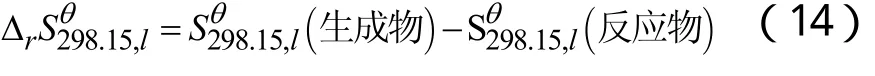
计算示例:以RA1反应为例进行化学反应热力学数据的计算。
25 ℃液相时:


其他反应的热力学数据估算值见表5。
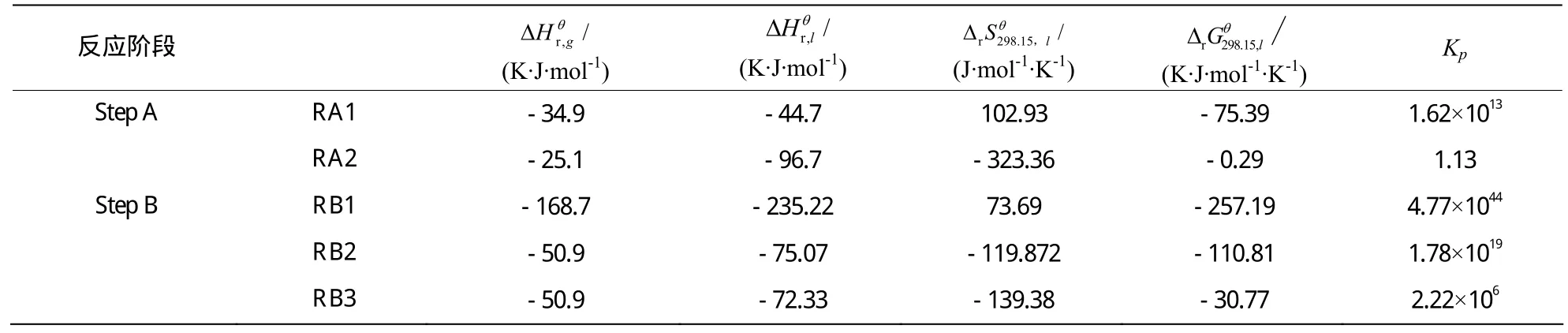
表5 反应热力学数据汇总表Table 5 Summary sheet of reaction thermodynamic data
3 甲基异丙基酮制备二甲基吡唑过程热力学分析
(1)A、B两个阶段的 5个反应均为放热反应且两个阶段的5个反应△G均小于零,表明5个反应均能自发进行。
(3)A阶段若提高目的产物的收率,应将反应体系中生成的水从体系中除去,使平衡向右移动。此阶段为放热反应,因此反应温度不宜过高。
(4)B阶段主反应 RB1和副反应 RB2、RB3均能自发进行到底。主反应RB1自发进行的趋势远远大于副反应RB2、RB3自发进行的趋势。但RB2的自发进行趋势也较大,因此,若要提高该阶段目的产物的收率需要加入催化剂,提高生成目的产物的选择性,可以选用碘或碘化物为催化剂。
(5)B阶段主反应 RB1放热较大,副反应放热不大。若提高目的产物的收率,此反应阶段反应温度不宜过高且不应高于反应物腙的常沸点。其原因是:①主反应RB1放热较大,温度过高反应不宜控制;②高于腙的常沸点后,大量腙进入气相,导致收率降低。
4 结 论
(1)本文计算的甲基异丙基酮制备3,4-二甲基吡唑反应的小于零, 远远大于1,说明该反应能自发进行到底。
(2)本文给基础物性热力学数据和热力学数据用于常沸点、临界参数的计算具有一定实用性及可靠性,计算值和文献值相差仅为均控制在3%以内。
(3)本文给出的基本物性数据和热力学数据简洁、实用,对于理论研究、工程设计、放大与生产具有参考价值。
(4)本研究对甲基异丙基酮合成3,4-二甲基吡唑的实验研究具有指导意义。
[1] 陈钟秀,顾飞燕,胡望明.化工热力学[M].第二版.北京:化学工业出版社,2001:270-290.
[2] 童景山.流体的热物理性质[M].北京:中国石化出版社,1996.
[3] 马沛生,化工数据[M].北京:中国石化出版社,2003:28-178.
[4] 王正烈,周亚平.物理化学 [M].第四版.北京:高等教育出版社,2001:114-123.
[5] 罗渝然. 化学键能数据手册[M]. 北京:科学出版社,2005:135-160.
Thermodynamic Calculation and Analysis on Preparation of 3,4-dimethyl-1H-yl-pyrazole From 3-Methyl-2-butanone
WANG Sha-sha,GUO Wa-li,TIAN Xu,FENG Jian,LI Ping,LIU Si-le,WANG Xiu-gang
(Shenyang University of Chemical Technology,Liaoning Shenyang 110142,China)
Joback group contribution method, Lydersen method, Riedel method, Ednister method, Rowlinson-Bondi corresponding state method and Benson method were used to estimate basic physical parameters and thermochemical property parameters related to the system of preparation of 3,4-dimethyl-1H-yl-pyrazole from 3-methyl-2-butanone ,such as boiling point , critical parameters ,eccentricity factor and gas heat capacity, liquid heat capacity ,latent heat of the evaporation, standard molar entropy of gas, entropy of phase transition. The bond energy method was used to calculate the standard reaction heat of the gas,at the same time the reaction heat under the actual reaction conditions was calculated as well as Gibbs free energy and chemical equilibrium constant, and characteristics of the reaction system were analyzed. These estimated data can provide the reference for theory research, engineering design and amplification of the reaction system.
Thermodynamic analysis; Group contribution; Hess law; 3,4-dimethyl-1H-yl-pyrazole
TQ 013.1
A
1671-0460(2012)01-0098-05
辽宁省教育厅创新团队项目(2008T159)。
2011-00-00
王莎莎 (1986-),女,辽宁沈阳人,硕士研究生,从事化工过程与产品开发。E-mail:594024255@163.com。
郭瓦力(1952-),女,教授,从事先进能源技术及化工过程与产品开发。E-mail:gwl3730@yahoo.com.cn。
猜你喜欢
能源化工(2021年3期)2021-12-31
今日农业(2021年2期)2021-11-27
今日农业(2020年23期)2020-12-31
电机与控制学报(2019年6期)2019-07-22
中国测试(2018年10期)2018-11-17
山东工业技术(2016年16期)2016-08-22
化工生产与技术(2016年5期)2016-03-13
合成化学(2015年1期)2016-01-17
中国塑料(2015年12期)2015-10-16
长江大学学报(自科版)(2014年1期)2014-03-20
