
程序升温分析谱图新模型
2012-11-06 05:12:42朱慧红孙素华伊晓东方维平
当代化工 2012年2期
金 浩,刘 杰,朱慧红,孙素华,杨 光,王 刚,伊晓东,方维平
(1. 中国石化抚顺石油化工研究院, 辽宁 抚顺 113001;2. 固体表面物理化学国家重点实验室, 醇醚酯化工清洁生产国家工程实验室, 厦门大学化学化工学院, 福建 厦门 361005)
模拟与计算
程序升温分析谱图新模型
金 浩1,刘 杰1,朱慧红1,孙素华1,杨 光1,王 刚1,伊晓东2,方维平2
(1. 中国石化抚顺石油化工研究院, 辽宁 抚顺 113001;2. 固体表面物理化学国家重点实验室, 醇醚酯化工清洁生产国家工程实验室, 厦门大学化学化工学院, 福建 厦门 361005)
以表面作用包括表面吸/脱附以及表面反应本征速率方程为基础,提出新的程序升温分析技术(TPAT)的数学模型。与经典的理论模型相比,这种新的TPAT理论模型更接近实际的程序升温分析实验过程。设计和进行特定催化剂的TPD、TPR及TPO实验,得到相应的程序升温谱图,采用新的TPAT理论模型模拟上述谱图,计算出相应的表面作用活化能等重要热力学参数。结果表明,新的理论模型具有良好的模拟性能,平均相对误差(ARD)小于1%。
程序升温分析技术;TPD;TPR;TPO;活化能;模型
在多相催化过程中,反应分子在催化剂表面的吸附、表面反应和脱附是三个最主要的步骤[1]。因此,为了探明其作用本质,最好是应用在反应或者接近反应条件下的动态分析技术[2]。而程序升温分析技术(TPAT)是一种重要的手段,它具有准确、简便和快速等特点,在多相催化研究中倍受重视,得到广泛研究和应用[3]。TPAT技术包括程序升温脱附(TPD)、程序升温还原(TPR)及程序升温氧化(TPO)等。
至今为止,对TPAT谱图所提供的信息利用率不高[4,5]。了解催化剂表面上存在几种不同的表面作用中心固然重要,但知道表面作用的强弱(由作用活化能衡量),对于催化剂研究更为重要。在实际应用中,一方面由于催化剂表面性质的非均匀性,谱图变的复杂,另一方面由于缺乏有效的理论处理手段,因而难以对谱图进行定量分析。虽然李湘等[6-7]早就开始此类研究,但对气相反应物分子在催化剂上作用强弱研究甚少或者不够深入,而且模型的模拟精度有待提高。因此,有必要探求新的、更为有效地处理TPAT谱图的方法。本文在表面作用本征速率方程基础上提出一种新的数学模型,该模型可以精确拟合各类TPAT谱图,并能计算出相应的表面作用活化能及相对谱峰面积等重要信息。
1 实验部分
1.1 催化剂制备
实验所用催化剂的制备如下:在一定配比的Mo-Ni-P浸渍液中添加10%柠檬酸,充分溶解,静置30 min左右,然后加入γ-Al2O3载体浸渍4 h,去除过量的浸渍液后,在低于100 ℃下充分干燥8 h,即得到实验所用的MoNiP/γ-Al2O3催化剂,记为HDS-1。
1.2 催化剂表征
H2-TPD 测试在Micromeritics Auto Chem 2920型全自动反应系统上进行。H2-TPR测试和O2-TPO测试在自建的装置上进行。使用上海海欣气相色谱仪在线检测还原过程中尾气中各组分的含量及其变化。
2 TPAT新模型
在TPAT过程中,总是存在某一随时间减小的物理量y,如在H2-TPD中,催化剂吸附氢量随时间减小;在H2-TPR中,气相H2浓度随时间降低。而TPAT谱图一般以正峰曲线出现,可将此曲线看成y的变化(减小)速率r'。y的变化本来是一种表面作用,可视为一种广义化学反应,进一步假设此“反应”级数为1。因此,借用广义质量作用定律,有


利用此式可拟合各种TPAT谱图,每1个峰拟合1次。
3 新模型应用
3.1 H2-TPD曲线模拟与讨论
程序升温脱附(TPD) 技术已广泛用于定性评价催化剂。这是一种等速升温条件下的程序升温过程。升温过程中,吸附质在催化剂表面脱附,导致气相组分浓度随时间而变,把这种变化过程纪录下来就得到TPD谱图。通常可通过对TPD 谱图的比较,定性分析催化剂对反应物分子的吸附性能。脱附活化能是脱附过程的一个重要热力学参数,它反映了反应物分子从催化剂表面解吸的难易程度。对于一般的负载型金属催化剂或多组分金属催化剂,还能反映其金属与载体或者各金属组分之间的分散度及其相互作用的强弱[8,9]。不同位置的峰,代表催化剂表面具有不同的吸附位,或者说反应物分子具有不同的吸附类型,也就具有不同的脱附活化能。而脱附峰相对面积的大小,可以衡量催化剂表面吸附分子的数量,后者往往与催化剂活性相关联。
图1是催化剂HDS-1的H2-TPD谱图(曲线yexp)。由图中可看出有两个脱附峰出现,说明H2以两种状态吸附于载体表面,催化剂具有两种不同的吸附中心[8]。第1个较小的低温峰出现在200 ℃左右,氢气吸附较弱、易脱附,且峰面积小,表明氢脱附量较少;第2个高温峰出现在490 ℃左右,说明此部分氢在催化剂表面上发生强吸附,或者说此类氢的脱附活化能较大,因此吸附较强、难脱附,且峰面积大代表氢脱附量较多。
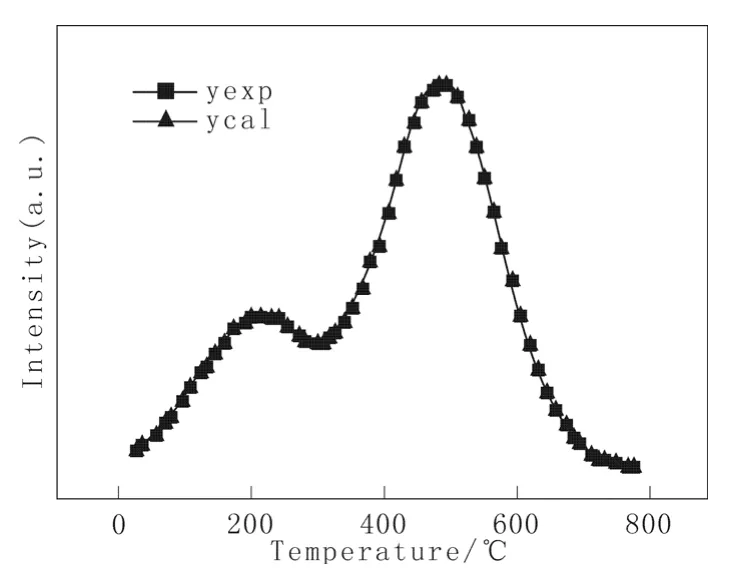
图1 催化剂HDS-1的H2-TPD谱图Fig.1 H2-TPD profiles of catalyst HDS-1
根据式(27)得到该催化剂的拟合脱附谱图(曲线ycal),同列于图1,计算参量列于表1。

表1 H2-TPD曲线的计算参量Table 1 Calculation Parameters of H2-TPD profiles
由图1和表1可见,模型的拟合精度好,总平均相对误差为0.62%;同时得到两个脱附峰对应的活化能,分别为14.988 kJ/mol和24.072 kJ/mol。高温脱附峰对应的活化能较低温峰的高许多,说明高温峰对应的脱附氢与催化剂作用更强,因而更不易脱附。两个脱附峰对应的相对谱峰面积可运用数值计算方法求得,分别为99.78和273.78,说明高温脱附峰对应的吸附氢量更大。这些数据较单纯的谱图更能形象具体的描述催化剂的活化能和氢脱附量等重要信息。
3.2 TPR/O曲线模拟与讨论
TPR/O实验与TPD相似,它用来表征氧化物或还原物催化剂的可还原程度或氧化程度,还可以通过TPR/O法研究得到金属催化剂各组分之间的相互作用强弱及组分种类等信息[10-12]。
该动力学模型应用到TPR/O,同样可以得到较好的模拟结果。图2为催化剂HDS-1的H2-TPR谱图(曲线yexp)。
根据式(27)得到该催化剂的拟合H2-TPR谱图(曲线ycal),同列于图2,计算参量列于表2。
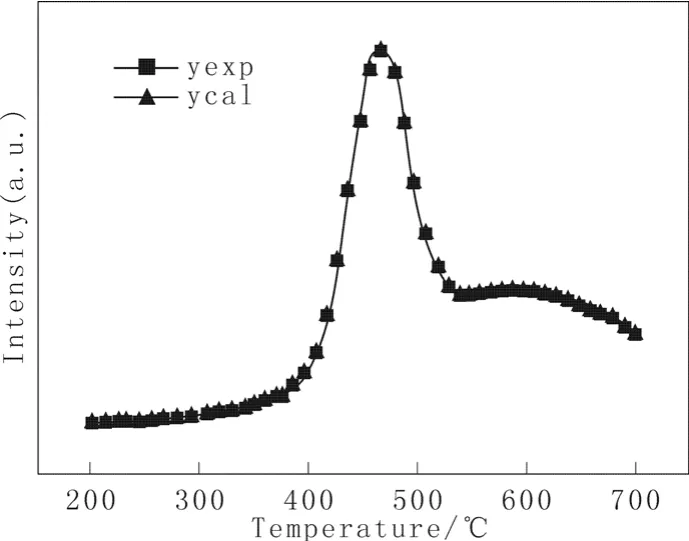
图2 催化剂HDS-1的H2-TPR谱图Fig.2 H2-TPR profiles of catalyst HDS-1

表2 H2-TPR曲线的计算参量Table 2 Calculation Parameters of H2-TPR profiles
由图2和表2可见,模拟结果与实验结果吻合十分好,根据式(27)修正后的动力学方程,总平均相对误差为0.11%。由图中可看出有两个还原峰:第一个低温还原峰出现在470 ℃左右,对应的活化能为22.028 kJ/mol,相对谱峰面积为29.1;第二个高温还原峰出现在590 ℃左右,对应的活化能为25.889 kJ/mol,相对谱峰面积为63.3。
图3为催化剂HDS-1的O2-TPO谱图(曲线yexp),根据式(27)得到该催化剂的拟合O2-TPO谱图(曲线ycal),同列于图3,计算参量列于表3。
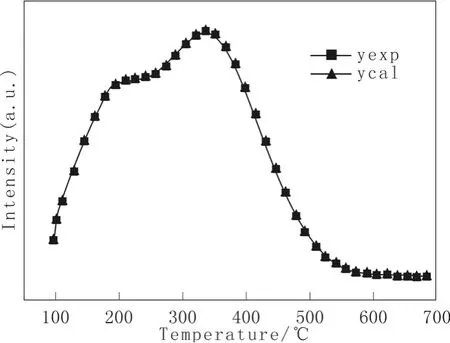
图3 催化剂HDS-1的O2-TPO谱图Fig.3 O2-TPO profiles of catalyst HDS-1

表3 O2-TPO曲线的计算参量Table 3 Calculation Parameters of O2-TPO profiles
由图3和表3可见,模拟结果与实验结果也十分吻合,根据式(27)修正后的动力学方程,总平均相对误差为0.75%。由图中可看出有两个氧化峰:第一个低温氧化峰出现在220 ℃左右,对应的活化能为14.99 kJ/mol,相对谱峰面积为299.6;第二个高温氧化峰出现在350 ℃左右,对应的活化能为19.76 kJ/mol,相对谱峰面积为785.9。
4 结 论
在表面作用包括表面吸/脱附以及表面化学反应本征速率方程基础上,提出一种新的TPAT数学模型。此模型可以精确地拟合TPAT实验谱峰,并且计算出相应的表面作用活化能等重要热力学参数。此模型的提出可促进TPAT技术的深入研究。
[1] Guo Y, Sakurai M, Kameyama H. Temperature programmed desorption/surface-reaction study of an anodic alumina supported Ag catalyst for selective catalytic reduction of nitric oxide with propene[J]. Applied Catalysis B: Environmental, 2008, 79(4): 382-393.
[2] Dias C R, Zãvoianua R, Portela M F. Study of the acid-base properties of SiO2-supported NiMoO4catalysts by temperature-programmed desorption: effect of the support[J]. Reaction Kinetics and Catalysis Letters, 2002, 77(2): 317-324.
[3] Castano P, Pawelec B, Fierro J L G, et al. Enhancement of pyrolysis gasoline hydrogenation over Pd-promoted Ni/SiO2-Al2O3catalysts[J]. Fuel, 2007, 86(15): 2262-2274.
[4]杨上闰. 二级脱附反应TPD谱图剖析[J]. 催化学报, 1985, 6(2): 179-182.
[5]王建国,李永旺,陈诵英,彭少逸. 沸石分子筛上程序升温脱附谱的M on te Carlo 模拟研究Ⅱ. 不同晶粒的沸石分子筛[J]. 催化学报, 1996, 17(1): 72-75.
[6]李湘, 李忠, 罗灵爱. 程序升温脱附活化能估算新模型[J]. 化工学报, 2006, 57(2): 258-262.
[7] Bhatia S, Beltramini J, Do D D. Temperature programmed analysis and its applications in catalytic systems[J]. Catalysis Today, 1990, 7(3): 309-438.
[8] Znak L, Zielin ski J. Effects of support on hydrogen adsorption/desorption on nickel[J]. Applied Catalysis A: General, 2008, 334(1-2): 268-276.
[9] Zheng J, Guo M, Song C S. Characterization of Pd catalysts supported on USY zeolites with different SiO2/Al2O3ratios for the hydrogenation of naphthalene in the presence of benzothiophene[J]. Fuel Processing Technology, 2008, 89(4): 467-474.
[10] Infantes-Molina A, Mé rida-Robles J, Rodrí guez-Castelló n E, et al. Effect of molybdenum and tungsten on Co/MSU as hydrogenation catalysts[J]. Journal of Catalysis, 2006, 240(2): 258-267.
[11] Boskovic G, Putanov P, Foettinger K, et al. Activation of Mo-based catalyst for paraffins isomerization[J]. Applied Catalysis A: General, 2007, 317(2): 175-182.
[12] Sundaramurthy V, Dalai A K, Adjaye J. The effect of phosphorus on hydrotreating property of NiMo/γ-Al2O3nitride catalyst[J]. Applied Catalysis A: General, 2008, 335(2): 204-210.
A New Mathematics Model of the Temperature Programmed Analysis Technology
JIN Hao1, LIU Jie1, ZHU Hui-hong1, SUN Su-hua1, YANG Guang,WANG Gang1, YI Xiao-dong2, FANG Wei-ping2
(1. Liaoning Fushun Research Institute of Petroleum and Petrochemicals,Liaoning Fushun 113001,China;
2. State Key Laboratory for Physical Chemistry of the Solid Surfaces,Engineering Laboratory for Green Chemical Productions of Alcohols,Ethers and Esters,College of Chemistry and Chemical Engineering,Xiamen University,Fujian Xiamen 361005,China)
Based on the surface effect including surface adsorption/desorption and intrinsic kinetics rate equation, a new mathematical model of the temperature programmed analysis technology was proposed. This model is different from the classical TPAT theory models, which indicates much more coincidence with the actual reactions than other models. TPAT (TPD, TPR and TPO) experiments were designed and carried out to receive their profiles. Based on these profiles of TPAT and the simulated experiments, the novel theory model was designed and the thermodynamics parameters (such as activation energy,etc.) were deduced and calculated by this model. The results show that this model has excellent simulation with the actual experiments, and the average relative errors are easily controlled less than 1%.
Temperature programmed analysis technology; TPD; TPR; TPO; Activation energy; Model
TQ 028
A
1671-0460(2012)02-0177-04
2011-00-00
金浩(1981-),男,辽宁锦州人,工程师,博士,2010年毕业于厦门大学物理化学专业,研究方向:从事渣油加氢催化剂的开发研制工作。E-m ail:jinhao.fshy@sinopec.com,电话:024-56389744。
猜你喜欢
探测与控制学报(2023年4期)2023-09-12 07:26:12
分析科学学报(2021年3期)2021-07-14 01:51:16
色谱(2021年6期)2021-05-06 02:18:56
科技资讯(2020年12期)2020-06-03 04:44:20
石油石化绿色低碳(2019年6期)2019-02-13 09:39:01
浙江大学学报(工学版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
中国资源综合利用(2016年4期)2016-01-22 08:27:23
物理实验(2015年9期)2015-02-28 17:36:51
数学年刊A辑(中文版)(2014年4期)2014-10-30 01:50:32
