
遗传性持续性胎儿血红蛋白增高症(HPFH)的分子机制
2012-09-19 03:52:12曾小红综述朱宝生校审
中国产前诊断杂志(电子版) 2012年2期
曾小红 综述 朱宝生校审
(昆明医科大学附属昆华医院 遗传诊断中心,云南 昆明 650032)
遗传性持续性胎儿血红蛋白增高症(HPFH)的分子机制
曾小红 综述 朱宝生*校审
(昆明医科大学附属昆华医院 遗传诊断中心,云南 昆明 650032)
遗传性持续性胎儿血红蛋白增高症(Hereditary persistence of fetal hemoglobin,HPFH)是成人红细胞中持续存在过量的胎儿血红蛋白 (Fetal hemoglobin,Hb F),血液学检查正常的遗传综合征。携带者常无临床症状。HPFH具有高度的遗传异质性,分子机制主要涉及11p15上β-类珠蛋白基因的遗传缺陷导致的Hb F异常高表达。最近的研究表明,HPFH具有数量性状遗传特点,其发生机制可能不局限于单纯的β-类珠蛋白基因上的遗传缺陷,HPFH还与多个基因座的异常有关,具有数量性状位点(quantitative trait loci,QTL)的遗传特征。主要包括QTL6q23和QTL2p15等的异常。通过HPFH来探索珠蛋白基因的网络化表达调控机制,为镰状细胞性贫血、重型地中海贫血等疾病的治疗研究开拓了新路径。
HPFH;分子机制;β-类珠蛋白基因;QTL
血红蛋白(Hemoglobin)是人体红细胞内的一种主要蛋白质,由珠蛋白和血红素结合而成,通过携氧释氧实现氧气在人体内的运输[1]。血红蛋白是由两条α珠蛋白链(αξ)和两条非α珠蛋白链(βγδε等)组成的四聚体。6种不同的珠蛋白肽链组合成人类的6种血红蛋白,按照其在人体内表达的先后顺序 分 别 是:Hb Gower1(ξ2ε2)、Hb Gower2(α2ε2)、Hb Portland(ξ2γ2)、Hb F(α2γ2)、Hb A(α2β2)、Hb A2(α2δ2);前3种为胚胎型血红蛋白,Hb F(Fetal hemoglobin,胎儿血红蛋白)为胎儿时期血红蛋白主要成分,出生后逐渐由Hb A取代,由于γ链有Gγ和Aγ两种亚型,所以Hb F也有两种构成:α2Gγ2和α2Aγ2。正常成人血红蛋白的主要成份是血红蛋白 A(Hb A,α2β2)占96.5%~97.5%;其余成分为:血红蛋白 A2(Hb A2,α2δ2),占2.5%~3.5%;胎儿红蛋白F(Hb F,α2γ2),占2%以下。当成人体内持续存在过量的Hb F时,就称为遗传性持续性胎儿血红蛋白增高症(Hereditary persistence fetal hemoglobin,HPFH)[2]。不同血红蛋白其携氧释氧能力不同,珠蛋白基因不同发育阶段特异性表达对维持机体的正常生理功能具有重要意义。珠蛋白基因不同发育阶段顺序性表达受复杂的网络系统精确调控,HPFH打破了正常珠蛋白基因开关机制的转换,从而吸引了研究者对其发生机制的探索,而HPFH也就成为了揭示珠蛋白基因顺序性表达调控的天然模型,还为地中海贫血(thalassemia,简称地贫)、镰状红细胞性贫血(Sickle cell disease,SCD)等病治疗的研究开拓新路径[1]。
HPFH首先于1955年在非洲人中发现。该病的分子病理存在着种族差异,故命名时一般冠以发现地或人种来修饰[3],而根据Hb F在细胞中的分布情况,又分为全细胞型HPFH和杂合细胞型HPFH[2]。β-类珠蛋白基因的遗传缺陷导致的 Hb F异常高表达一直以来都是HPFH分子机制的重要学说。HPFH患者红细胞中Hb F数量的连续变化,以及Hb F表达相关的多个数量性状位点(Quantitative trait loci,QTL)被确定,均表明HPFH具有数量性状遗传特性[4]。非缺失型HPFH的机制已经不再局限于11p15的β-类珠蛋白基因上,基因多态性与Hb F表达的数量性状位点学说成为了阐述HPFH机制的重要理论。
1 β-类珠蛋白基因的遗传缺陷导致HPFH
1.1 β珠蛋白基因簇内片段缺失 缺失型HPFH是指由于β珠蛋白基因簇内大片段DNA序列缺失引起的HPFH。β珠蛋白基因簇上有多个参与珠蛋白基因表达调控的元件,比如基因座控制区(locus control region,LCR)上的高敏感位点(hypersensitive site,HS)(结构示意见图1)、启动子 (promoter)、增 强 子 (enhancer)、沉 默 子(silencer)、以及其他与表达调控相关的基因序列,而β珠蛋白基因簇上大片段DNA序列的缺失扰乱了γ珠蛋白基因正常关闭和表达减速,从而产生了HPFH[5]。目前解释缺失型 HPFH具体机制的学说主要有以下3方面:①缺失片段包括珠蛋白表达调控中的高敏感位点3′HS-1,从而使其对γ基因表达的抑制作用丧失,位置效应导致γ基因受上游5′端的LCR调控,同时没有β-基因竞争使得γ链表达持续升高[6-9];②缺失使该基因断裂点3′端下游的增强子靠近γ-基因而产生更强的正效应,导致Hb F高表达,即“距离效应”[3];③γ-珠蛋白基因沉默子缺失,导致成人时期本应关闭的γ-珠蛋白基因持续表达[10-12]。除此之外,γ-珠蛋白基因3′端两个沉默元件Enh和F可能与Hb F表达调控相关,Maria等[13]通过转基因鼠、质粒构建、FISH等技术证实沉默元件Enh和F的缺失可以导致Hb F持续性高表达。缺失型HPFH常根据其发现人群,缺失片段的位置及大小来区分,常见的有7种(图2)[3,14],东南亚缺失型HPFH是报道较普遍的一种,有研究认为[15]其机制主要是 3′HS-1 缺失同时伴有 3′端HPFH-3增强子的存在。其他新缺失也不断有报道,Philippe Joly等[16]分别在法国籍、阿尔及利亚籍的Hb F升高患者中发现了3种新的缺失,分别命名为 French 缺失,French West-Indies缺失,Algeria缺失,其缺失片段长短及位置见图2[16],具体的机制现在尚未阐明。
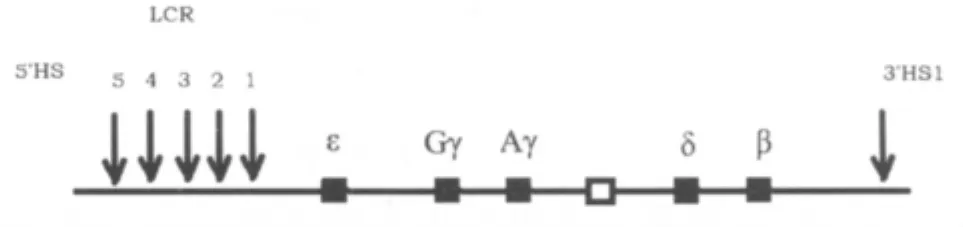
图1 人类β基因座控制区结构示意:↓为HS,
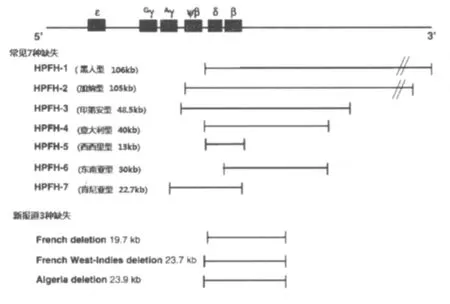
图2 几种缺失型HPFH的缺失片段长度及位置
1.2 Aγ基因和Gγ基因的点突变 目前发现的β珠蛋白基因簇上γ-基因(Gγ或Aγ)启动子区的点突变导致 的HPFH已不下19种[3,14,17](表1)。常见的有 Gγ基因上游区域-161,-175,-202和 Aγ基因上游区域的-177,-196和-198等位点的核苷酸取代[1]。与β基因启动子区点突变不同,γ基因(Gγ或Aγ)启动子区的点突变可使其与反式作用因子如 GATS-1、OTF-1、SP1等的结合能力下降[3],增强被调控基因的表达,从而使γ基因过度表达。同时突变还可以产生新的结合位点,增强转录激活物与其结合的能力,使得γ基因在成人体内高表达。有研究认为[18]蛋白 DNMT1、p52与 Aγ-198 (T→C)型 HPFH的形成有关,Aγ-198(T→C)突变改变了γ-基因对反式作用因子SP1的亲和力,同时,它还产生了γ基因表达不可缺少的CACCC盒[19]。与198(T→C)不同,Aγ195(C→G)突变是巴西人群中HPFH的常见原因,它不影响SP1,也不会产生新的CACCC盒,有研究发现[20]Aγ195(C→G)突变不能单独导致γ基因在成人体内高表达,当加入LCR(HS2)片段,才可观察到表达适度的增加,但其具体影响γ基因高表达的机制还有待进一步研究。在希腊人群中新发现 HPFH的两种突变[17]Gγ-196 C→T和Aγ-201C→T,XmnⅠ酶切位点被认为是导致 Hb F高表达的因素[3],但是否与 Gγ-158(C→T)产生的XmnⅠ酶切位点一样,要在某些压力作用下,比如纯合子的SCA和地贫,才能导致Hb F升高,还未明确。Aγ-196(C→T)突变是在中国人群中最早鉴定出的一种γ基因启动子区突变[3]。
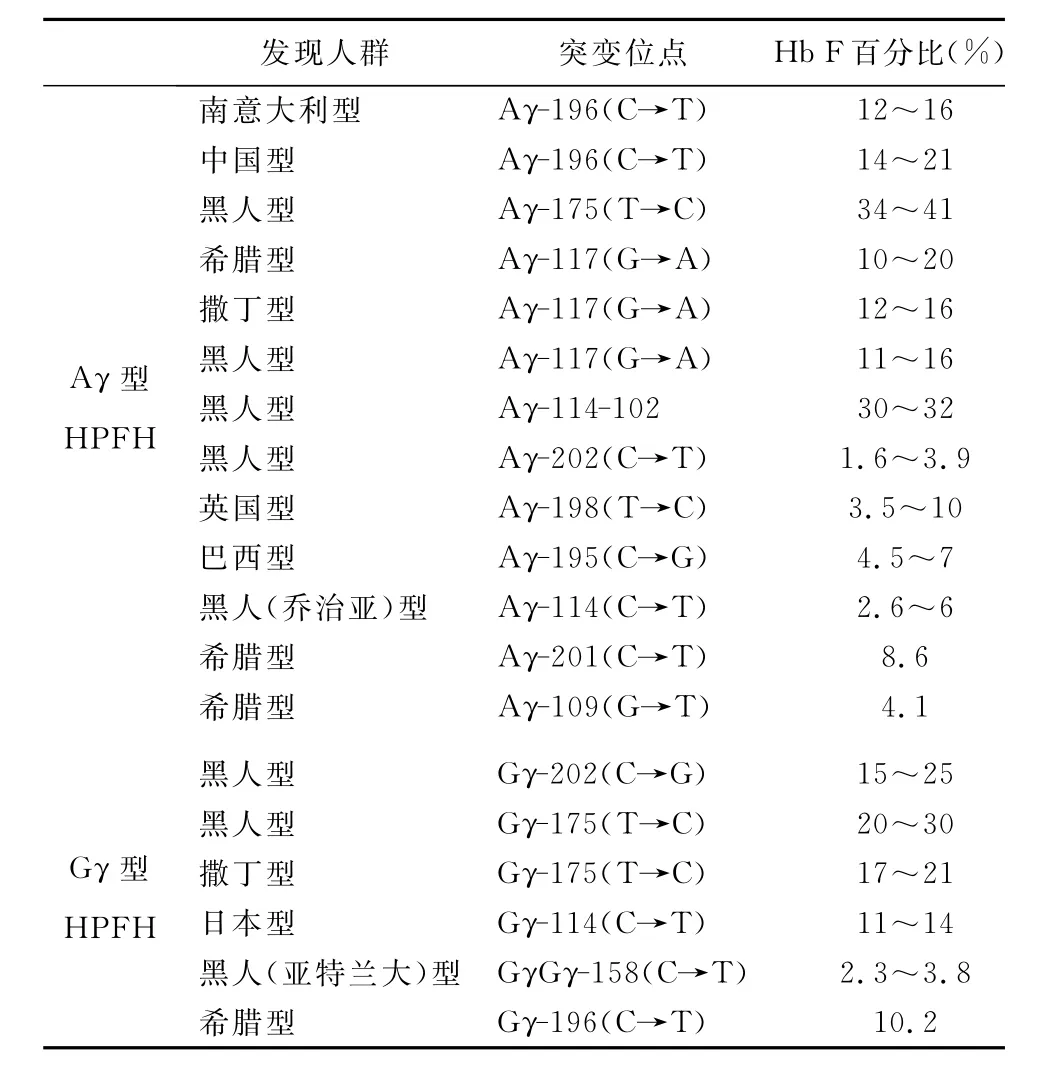
表1 β珠蛋白基因簇上γ-基因(Gγ或Aγ)启动子区的点突变
1.3 Gγ、Aγ及其它基因的多态性 β-类珠蛋白基因的遗传缺陷与HPFH相关性不仅仅表现在基因突变或者大片段的缺失,还与其基因序列上的一些多态性有关。Jouini等[21]通过对68名突尼斯正常成人和HPFH患者β-类珠蛋白基因上多态性区域进行分析表明:Gγ-158(C→T)多态位点,Gγ第二内含子 上 TG18CG2CACG、TG9TCAGTG2CG2、TG11CG4重复序列和β基因启动子序列上AC2AT6T9和AC2AT7T8重复序列与Hb F高表达具有相关性。
Gγ-158(C→T)在多个人群的HPFH携带者中均有发现,尽管该位点位于Gγ启动子区,但其并不影响转录因子的活性,而是产生了XmnⅠ酶切位点[22],只有在重型地贫[23]和镰状细胞性贫血患者体内[24],该突变才能导致 Hb F显著升高,正常人体内只会导致轻微的 Hb F增高[25]。因此,Gγ-158(C→T)也被认为是影响γ基因表达的一个多态位点,其发挥作用的机制有研究认为可能与8q12.1编码的某些调控因子或者亚基,对XmnI-Gγ位点起转录激活或抑制的作用有关[26]。
Gγ和Aγ基因的不同之处在于其第二内含子区域内(TG)n(CG)m 微卫星序列[27],该区域多态性是IVS2上所谓的“热点”区域基因重组的结果[28]。Jouini等[21]发现只有在 HPFH 患者中才有TG18CG2CACG、TG9TCAGTG2CG2和TG11CG4序列,推测(TG)n(CG)m多态性与 Hb F升高有相关性。而Lapoumeroulie等[27]发现在西西里中间型地贫患者中TG18CG2CACG序列有相当的出现频率,而在正常人群和重型地贫患者中该序列几乎完全缺如[29],两者结论都支持(TG)n(CG)m 多态性与Hb F升高有相关性这一观点。
有研究报道(AT)xTy重复序列可能通过产生蛋白BPⅠ抑制β-类珠蛋白基因来影响Hb F表达[30]。 在 Jouini 等[21]研 究 中 AC2AT6T9 和AC2AT7T8只在Hb F升高组中发现,因此推测β-类珠蛋白基因启动子区域内(AC)n(AT)xTy重复序列与Hb F高表达有相关性。同时还发现Hb F表达的多少与其所携带的多态位点的数目无明显相关性,多态位点之间可能是各自发挥作用,也有可能存在相互影响。
近年来基因多态性研究一直是HPFH机制研究的热点,目前发现的不管是位于β-类珠蛋白基因簇上的多态性还是其他与Hb F数量性状相关基因座上的多态性,其影响Hb F表达具体机制都有待进一步阐述。就β珠蛋白基因簇上多态性而言,有研究认为γ基因第二内含子(IVS2)上一些多态序列与参与珠蛋白基因表达调控的一些重要因子的基因有相互影响,比如 GATA-1[31]和 KLF-1[32]等,多态序列可通过降低某些转录激活因子的亲和力或者增加抑制因子的亲和力来影响Hb F表达[29]。
2 其他染色体上与HPFH相关的基因座
β-类珠蛋白基因的遗传缺陷不能完全解释所有HPFH的形成机制,有临床证据表明HPFH具有数量遗传的特性。针对这一特性,研究者运用GWAS等新的遗传学研究法发现了一些与HPFH形成相关的基因座,目前报道的除11P15上β-类珠蛋白基因以外与HbF表达相关的数量性状位点(Quantitative trait loci,QTL)至 少 有:2p15[33]、6q23[34]、Xp22.2[35]、8q12.1[26]、19p13.12-13[36]。报道较多的主要是QTL6q23和QTL2p15,而19p13.12-13是近年新报道的一个与HbF表达相关的数量性状位点。
2.1 QTL6q23 研究发现6q23上并未出现与Hb F增高有关的突变或缺失,其上1.5Mb区域有5个蛋白编码基因,ALDH8A1(Aldehyde dehydrogenase 8 family member A1)、HBS1L(HBS1-like protein)、MYB (v-MYB avian myeloblastosis viral oncogene homolog)、AHI1(Abelson helper integration site 1)和PDE7B (Phos-phodiesterase 7B),其 中,MYB、HBS1L在Hb F的高表达贡献最大[37]。GWAS研究表明,HBS1L第一个外显子的32位C32T的多态性显著影响地中海贫血的严重性和HbF含量[38]。而cMYB对保持细胞分化增殖的平衡有关键作用[39],尤其对红细胞分化成熟为有核红细胞产生成人血红蛋白的过程[40]。而且MYB还参与了珠蛋白基因转录信号传导,受到cAMP的调节而参与γ-球蛋白基因的表达。HBS1L-MYB基因间多态性(HBS1L-MYB intergenic polymorphism,HMIP)在北欧人群中被证实能影响Hb F含量变化[41]。
2.2 QTL2p15 2p15与Hb F含量相关性主要体现在该区域内的 BCL11A(B-cell CLL/lymphoma 11A)基因上。BCL11A基因最早发现与正常淋巴细胞发育有关,并参与造血肿瘤的发生。它由5个外显子和4个内含子组成,其第二个外显子区有2个单核苷酸多态性,分别是rs11886868(P=10-35)和rs1427407(P<10-19),而在北欧健康人群,撒丁尼亚地中海贫血患者和中国地中海贫血患者中都证实其与HbF含量的关联度最高,其中rs11886868(P=10-35)位点存在 C/T 多态性,正常碱基为C[41],而rs1427407(P<10-19)位点存在 G/T 多态性,正常的G转变为T可显著增加HbF的含量[42]。除此之外,研究还发现BCL11A基因表达与γ珠蛋白基因的表达负相关,HPFH患者体内BCL11A基因表达低于正常人[43],具体机制尚待进一步研究。
2.3 QTL19p13.12-1 Borg等[36]研究发现位于19p13.12-1上的KLF1基因对HPFH的形成有双向调节作用。一方面,其编码的KLF1是红细胞转录调节因子,直接作用于γ珠蛋白基因启动子区的调控序列,参与成人珠蛋白基因表达的调控;另一方面,KLF1是BCL11A基因的激活因子,而BCL11A基因是γ珠蛋白基因表达的负调控因子,因此当KLF1基因突变导致KLF1单倍剂量不足(Haploinsufficiency)时,BCL11A基因表达下降,相应地就会出现Hb F在成人体内高表达。目前在HPFH患者体内发现的KLF1基因突变有p.M39L和p.K288X两种。
3 小 结
HPFH本身无临床症状,携带者的血液学检查正常,不影响人体健康,对其分子机制的研究旨在探索珠蛋白基因表达调控的奥秘。在重型β地中海贫血(β-thalassemia major)和 镰 状 红 细 胞 性 贫 血(Sickle cell disease,SCD)患者体内,HPFH 的出现可以改善贫血症状;羟基脲(Hydroxyurea)作为减少SCD并发症的治疗药物主要就是通过增加Hb F表达,来减轻溶血对机体的损害[44]。而其增加Hb F表达主要是通过QTL6q23上MYB的调节作用实现,这无疑为其他HPFH机制研究的临床应用提供了参考。BCL11A基因作为重型地贫和SCD新治疗药物靶点的研究也不断有报道,KLF1基因突变可上调Hb F表达,为治疗严重血红蛋白病也带来成功的希望。总之,通过对HPFH的不同形成机制的研究,来探索珠蛋白基因的网络化表达调控机制,为镰状细胞性贫血、重型地中海贫血等疾病的治疗研究开拓了新路径。
[1]曾溢滔.人类血红蛋白[M].北京:科学出版社,2002.94-96.
[2]顾援朝.遗传性胎儿血红蛋白持续存在综合征[J].国外医学分子生物学分册,1980,4:167-169.
[3]徐湘民.见于中国人的HPFH和δβ-地中海贫血的分子基础[J].中华医学遗传学杂志,1998,15(05):315-317.
[4]Menzel S,Thein SL.Genetic architecture of hemoglobin F control[J].Curr Opin Hematol.2009,16(3):179-186.
[5]de Andrade TG, Peterson KR, Cunha AF,et al.Identification of novel candidate genes for globin regulation in erythroid cells containing large deletions of the human betaglobin gene cluster[J].Blood Cells Mol Dis,2006,37(2):82-90.
[6]Townes TM,Behringer RR.Human globin locus activating region:role in temporal control[J].Trends Genet,1990,6:219-223.
[7]Arcasoy MO,Romana M,Fabry ME,et al.High levels of human gamma-globin gene expression in adult mice carrying a transgene of deletion-type hereditary persistence of fetal hemoglobin[J].Mol Cell Biol,1997,17(4):2076-2078.
[8]Feingold EA,Penny LA,Nienhuis AW,et al.An olfactory receptor gene is located in the extended human beta-globin gene cluster and is expressed in erythroid cells[J].Genomics,1999,61(1):15-23.
[9]Katsantoni EZ,Langeveld A,Wai AW,et al.Persistent gamma-globin expression in adult transgenic mice is mediated by HPFH-2,HPFH-3,and HPFH-6breakpoint sequences[J].Blood,2003,102(9):3412-3419.
[10]T Huisman,W Schroeder,G Efremov,et al.The present status of the heterogeneity of fetal hemoglobin inβthalassemia:an attempt to unify some observations in thalassemia and related conditions[J].Ann N Y Acad Sci,1974,232:107-124.
[11]Vitale M,Calzolari R,Di Marzo R,et al.A region upstream of the human delta- globin gene shows a stage-specific interaction with globin promoters in erythroid cell lines[J].Blood Cells Mol Dis,2001,27(5):874-881.
[12]Calzolari R,McMorrow T,Yannoutsos N,et al.Deletion of a region that is a candidate for the difference between the deletion forms of hereditary persistence of fetal hemoglobin and deltabeta-thalassemia affects beta-but not gamma-globin gene expression[J].EMBO J,1999,18(4):949-958.
[13]Gazouli M,Katsantoni E,Kosteas T,et al.Persistent fetal gamma-globin expression in adult transgenic mice following deletion of two silencer elements located 3'to the human Agamma-globin gene[J].MOL MED,2009,15(11-12):415-424.
[14]Forget BG.Molecular basis of hereditary persistence of fetal hemoglobin[J].Ann N Y Acad Sci,1998,850:38-44.
[15]Changsri K, Akkarapathumwong V,Jamsai D,et al.Molecular mechanism of high hemoglobin F production in Southeast Asian-type hereditary persistence of fetal hemoglobin[J].Int J Hematol,2006,83(3):229-237.
[16]Joly P,Lacan P,Garcia C,et al.Identi cation and molecular characterization of four new large deletions in theβ-globin gene cluster[J].Blood Cells Mol Dis,2009,43(1):53-57.
[17]Tasiopoulou M,Boussiou M,Sinopoulou K,et al.G gamma-196C->T,A gamma-201C->T:two novel mutations in the promoter region of the gamma-globin genes associated with nondeletional hereditary persistence of fetal hemoglobin in Greece[J].Blood Cells Mol Dis,2008,40(3):320-322.
[18]Olave IA,Doneanu C,Fang X,et al.Purification and identification of proteins that bind to the hereditary persistence of fetal hemoglobin-198mutation in the gammaglobin gene promoter[J].J Biol Chem,2007,282(2):853-862.
[19]Li Q,Duan ZJ,Stamatoyannopoulos G.Analysis of the mechanism of action of non-deletion hereditary persistence of fetal hemoglobin mutants in transgenic mice[J].EMBO J,2001,20(1-2):157-164.
[20]Takahashi T,Schreiber R,Krieger JE,et al.Analysis of the mechanism of action of the Brazilian type(Agamma-195C->G)of hereditary persistence of fetal hemoglobin[J].Eur J Haematol,2003,71(6):418-424.
[21]Jouini L,Bibi A,Ouali ,et al.Contribution ofβ-globincluster polymorphisms to raise fetal hemoglobin levels in normal adults[J].Mol Biol Rep,2012,39(4):3443-3452.
[22]Labie D,Dunda-Belkhodja O,Rouabhi F,et al.The-158 site 50to the Gγgene and Gγexpression[J].Blood,1985,66(6):1463-1465.
[23]Camaschella C, Mazza U, Roetto A,et al. Genetic interactions in thalassemia intermedia:analysis of betamutations,alpha-genotype,gamma-promoters,and beta-LCR hypersensitive sites 2and 4in Italian patients[J].Am J Hematol,1995,48(2):82-87.
[24]Lettre G,Sankaran VG, Bezerra MA,et al.DNA polymorphisms at the BCL11A,HBS1L-MYB,andβ-globin loci associate with fetal hemoglobin levels and pain crises in sickle cell disease[J].Proc Natl Acad Sci U S A,2008,105(33):11869-11874.
[25]Sampietro M,Thein SL,Contreras M,et al.Variation of Hb F and F-cell number with the Gγ Xmn I (C-T)polymorphism in normal individuals[J].Blood,1992,79(3):832-833.
[26]Garner CP,Tatu T,Best S,Creary L,et al.Evidence of Genetic Interaction between the β-Globin Complex and Chromosome 8q in the Expression of Fetal Hemoglobin[J].Am J Hum Genet,2002,70(3):793-799.
[27]Lapoumeroulie C,Castiglia L,Ruberto C,et al.Genetic variations in human fetal globin gene microsatellites and their functional relevance[J].Hum Genet,1999,104(4):307-314.
[28]Slighton JL,Blechl AE,Smithies O.Human fetal Gγand Aγ-globin genes:complete nucleotide sequences suggest that DNA can be exchanged between these duplicated genes[J].Cell,1980,21(3):627-638.
[29]Zertal-Zidani S,Ducrocq R,Sahbatou M,et al.Foetal haemoglobin in normal healthy adults:relationship with polymorphic sequences cis to the beta globin gene[J].Eur J Hum Genet,2002,10(5):320-326.
[30]Elion J,Berg PE,Lapoumeroulie C,et al.DNA sequence variation in a negative control region 50to theβ-globin gene correlates with the phenotypic expression of theβSmutation[J].Blood,1992,79(3):787-792.
[31]Seshasayee D,Geiger JN,Gaines,et al.Intron 1elements promote erythroid-specific GATA-1gene expression[J].J Biol Chem ,2000,275(30):22969-22977.
[32]Asano H,Li XS,Stamatoyannopoulos G.FKLF-2:a novel Kruppel-like transcriptional factor that activates globin and other erythroid lineage genes[J].Blood,2000,95(11):3578-3584.
[33]Menzel S,Garner C,Gut I,et al.A QTL influencing F cell production maps to a gene encoding a zinc-finger protein on chromosome 2p15[J].Nat Genet,2007,39(10):1197-1199.
[34]Craig JE,Rochette J,Fisher CA,et al.Dissecting the loci controlling fetal haemoglobin production on chromosomes 11p and 6q by the regressive approach[J].Nat Genet,1996,12(1):58-64.
[35]Dover GJ,Smith KD,Chang YC,et al.Fetal hemoglobin levels in sickle cell disease and normal individuals are partially controlled by an X-linked gene located at Xp22.2[J].Blood,1992,80(3):816-824.
[36]Borg J, Papadopoulos P, Georgitsi M, et al.Haploinsufficiency for the erythroid transcription factor KLF1causes hereditary persistence of fetal hemoglobin[J].Nat Genet,2010,42(9):801-805.
[37]Close J,Game L,Clark B,et al.Genome annotation of a 1.5Mb region of human chromosome 6q23encompassing a quantitative trait locus for fetal hemoglobin expression in adults[J].BMC Genomics,2004,5(1):33.
[38]Pandit RA,Svasti S,Sripichai O,et al.Association of SNP in exon 1of HBS1Lwith hemoglobin F level in beta0-thalassemia/hemoglobin E[J].Int J Hematol,2008,88(4):357-361.
[39]Kuroyanagi Y, Kaneko Y, Muta K,et al.cAMP differentially regulates gamma-globin gene expression in erythroleukemic cells and primary erythroblasts through c-Myb expression[J].Biochem Biophys Res Commun,2006,344(3):1038-1047.
[40]Iliadou A,Evans DM,Zhu G,et al.Genomewide scans of red cell indices suggest linkage on chromosome 6q23[J].J Med Genet,2007,44(1):24-30.
[41]Thein SL,Menzel S,Peng X,et al.Intergenic variants of HBS1L-MYB are responsible for a major quantitative trait locus on chromosome 6q23influencing fetal hemoglobin levels in adults[J].Proc Natl Acad Sci U S A,2007,104(27):11346-11351.
[42]Uda M, Galanello R, Sanna S,et al.Genome-wide association study shows BCL11Aassociated with persistent fetal hemoglobin and amelioration of the phenotype of betathalassemia[J].Proc Natl Acad Sci U S A,2008,105(5):1620-1625.
[43]Sankaran VG, Menne TF,Xu J,et al.Human fetal hemoglobin expression is regulated by the developmental stage-specific repressor BCL11A[J].Science,2008,322(5909):1839-1842.
[44]Ma Q,Wyszynski DF,Farrell JJ,et al.Fetal hemoglobin in sickle cell anemia:genetic determinants of response to hydroxyurea[J].Pharmacogenomics J,2007,7(6):386-394.
编辑:王腾
Molecular Basis of the Hereditary Persistence of Fetal Hemoglobin(HPFH)
Zeng Xiao-hong,Zhu Bao-sheng*.
(Genetic Diagnosis Center,Affiliated Kunhua Hospital of Kunming Medical college,Kunming 650032,China)
Hereditary persistence of fetal hemoglobin(HPFH)is a group of genetic syndromes with normal blood test and the persistence of excessive Hb F(Fetal hemoglobin)in the adult red blood cells.Most carriers of HPFH have no clinical symptoms.HPFH has a high degree of genetic heterogeneity.The molecular mechanisms of HPFH involve in abnormal expression of Hb F caused by genetic defects of betaglobin gene cluster on 11p15.Recently,several studies revealed that HPFH has characteristics of quantitative trait loci(QTL).The mechanism of HPFH may not only be limited to genetic defects within the beta-globin gene cluster,but also be related to abnormalities in multiple loci,including QTL6q23and QTL2p15.Through exploring the expression and regulation mechanisms of globin gene regulation network,a new gate for studies on the treatment of sickle cell anemia and thalassemia has been opened.
HPFH;molecular mechanism;beta-globin gene;quantitative trait loci
R394.3
A
2012-05-11)
本文受云南省科技厅-昆明医学院联合专项重点项目“云南地中海贫血遗传异质性与防治对策研究”(项目编号:2011FB164)资助
*通讯作者:朱宝生,Email:bszhu@yahoo.cn
猜你喜欢
世界科学技术-中医药现代化(2022年3期)2022-08-22 00:33:26
检验医学与临床(2021年14期)2021-07-29 07:40:24
西北农林科技大学学报(自然科学版)(2019年8期)2019-07-17 02:43:32
西南农业学报(2016年6期)2016-04-16 05:12:47
法医学杂志(2015年4期)2016-01-06 12:36:36
检验医学与临床(2015年14期)2015-03-16 01:46:53
现代检验医学杂志(2015年1期)2015-02-06 01:59:18
遗传(2015年5期)2015-02-04 03:06:55
海洋科学(2014年12期)2014-12-15 03:35:00
河南医学研究(2014年7期)2014-02-27 14:53:42
