
紫球藻多糖化学结构解析
2012-09-15 04:25孙利芹马翠华王长海
天然产物研究与开发 2012年5期
孙利芹,王 凌,马翠华,王长海
1烟台大学生命科学学院,烟台 264005;2大连理工大学环境与生物工程系,大连 116024
紫球藻多糖化学结构解析
孙利芹1,2*,王 凌1,马翠华1,王长海2
1烟台大学生命科学学院,烟台 264005;2大连理工大学环境与生物工程系,大连 116024
为明确紫球藻多糖的化学结构,本文采用化学分析和光谱分析方法对紫球藻多糖的一级糖链结构进行了分析。GC分析表明该多糖由木糖、葡萄糖和半乳糖组成,为一种杂多糖,其摩尔比为:2.96∶1.25∶3.06;红外光谱分析结果显示紫球藻多糖为硫酸化多糖,糖苷键类型为β构型;化学分析结果推断紫球藻多糖糖链连接方式以1→3为主,存在少量1→2,1→4,1→6键型,且半乳糖在支链或主链末端有较大量的存在,木糖和葡萄糖在主链或靠近主链区域有特定分布;NMR分析显示紫球藻多糖的硫酸酯基连在C-6上,且多糖的糖苷键为β型; GC-MS联机分析进一步确定紫球藻多糖为一种主要含有1→3糖苷键,并含有1→4,1→6糖苷键的杂多糖。综合上述分析,推断出紫球藻多糖的糖链主链的重复单元结构。
紫球藻;多糖;糖链结构;化学分析;光谱分析
多糖的生物活性与其空间构象、分子量大小、分支度和溶解度有密切关系,化学结构的微小变化可能对它的构象和多糖的特性产生较大的影响[1]。近十几年,研究者已经得到了一系列多糖重复单元的结构,这些多糖的重复结构单元结构通过酸水解、甲基化分析、高碘酸氧化、Smith降解、乙酰解、酶解和1D、2D核磁共振谱等方法被解析[2-4]。但是由于多糖的结构变化多样、糖链结构的复杂性,糖链结构分析一直是多糖结构测定的难点。
紫球藻(Porphyridium Cruentum)是目前发现的红藻门中唯一的单细胞藻类,细胞生长过程中会大量合成并向胞外分泌一种磺酸化多糖,其分子量达2~7×106Da[5]。近几年,紫球藻多糖的抗病毒、抗氧化、抗辐射和降低血酯等生物活性相继被报道[6-9],然而,对紫球藻多糖糖链结构的研究目前除了Gloaguen[4]采用NMR分析了紫球藻多糖降解后的一种寡糖片段的糖链连接方式外,其余的报道仅限于对糖链单糖组成的分析[4-5,10]。
本研究主要通过化学分析和光谱分析方法,对紫球藻多糖的单糖组成、单糖残基间的连接测序、糖苷键构型及重复单元结构等糖链的一级结构进行分析,旨在为紫球藻多糖高级结构的研究及多糖结构与功能的构效关系研究奠定基础。
1 实验部分
1.1 仪器与试剂
1.1.1 实验材料
紫球藻多糖(EPS),由烟台大学海洋生化工程研究室提取、纯化,经冷冻干燥后获得的白色絮状固体。
D-甘露糖、D-半乳糖、D-木糖、葡萄糖醛酸AR,Sigma公司;L-岩藻糖、L-鼠李糖,AR,Treechem公司;肌醇,AR,中国医药集团上海化学试剂公司;其它试剂均为国产分析纯。
1.1.2主要仪器
Shimadzu IR-400型红外光谱仪(日本岛津株式会社)、Agilent6820型气相色谱仪(美国安捷伦科技有限公司)、Agilent1100型高效液相色谱仪(美国安捷伦科技有限公司)、MS-GC2010气质联用机(日本岛津株式会社)、Bruker AVANCE 400 Digital NMR核磁共振仪(德国Bruker公司)。
1.2 实验方法
1.2.1 单糖组成分析[11]
衍生物的制备—糖醇乙酸酯,以肌醇为内标物,具体方法参见文献[11]。将标准单糖制成糖醇乙酸酯衍生物,直接进行气相色谱分析,记录标准单糖的保留时间(RT)。
样品的测定:多糖样品20 mg,加入8 mL 2 mol/ L的三氟乙酸,封瓶,120℃下置入高压灭菌锅水解2.0 h,水解液于60℃减压蒸发至干,105℃烘箱中烘至恒重。称重,得水解后单糖质量,按上述衍生物的制备方法加入各药品及试剂,产物作气相色谱分析。记录样品中单糖的保留时间,并与标准单糖的气相色谱图进行比较,确定紫球藻胞外多糖的单糖组成。
气相色谱分析条件:色谱柱:HP-5MS毛细管柱(30 m ×0.32 mm ×0.5 μm)、氢火焰离子化检测器;程序升温:初始温度200℃保持2 min,以20℃/ min升温至240℃,保持2 min,再以5℃/min升温至250℃,保持1 min;进样口温度250℃,进样量1 μL,以氮气为载气,流速2 mL/min。
1.2.2 红外光谱分析
取干燥的紫球藻多糖EPS0 1.0 mg,以KBr压片,在4000~400 cm-1的范围内进行红外光谱扫描,记录红外光谱图。
1.2.3 部分酸水解[11]
分别将30 mg紫球藻多糖与0.05、0.5和1 mol/L的三氟乙酸(TFA)3 mL混合,封管,于75℃水解16 h。用截留分子量为8000 Da的透析袋将水解片断分级,分别得到袋外上清、袋内上清袋内沉淀三种组分。
各组分适当浓缩、冻干后,按内标法定量,GC分析各单糖相对含量,气相色谱分析条件同1.2.1。
1.2.4 高碘酸氧化和Smith降解[11]
1.2.4.1 准确称取糖样50 mg,用少量水溶解,加入50 mL容量瓶中;准确称取160.4 mg NaIO4,用少量水溶解后,加入容量瓶中,定容,使NaIO4终浓度为15 mmol/L。
1.2.4.2 放置在暗处反应(室温),间隔时间(0、6、12、24、36、……,78 h)取样0.l mL,蒸馏水稀释后用紫外分光光度计,以蒸馏水作空白对照,在223 nm波长处测光密度值,直到光密度值恒定为止。
1.2.4.3 加乙二醇终止反应,高碘酸氧化完成。
1.2.4.4 甲酸测定:取4 mL上述氧化液,加1滴溴甲酚(红)紫作指示剂,用0.05 mol/L的NaOH(用邻苯二甲酸氢钾标定)滴定,计算得甲酸生成量。
1.2.4.5 将乙二醇处理后的溶液用蒸馏水透析48 h,于40℃以下减压浓缩至小体积,加入硼氢化钠以还原多糖醛,于室温暗处搅拌24 h,用0.1 mol/L乙酸调pH至5.5,以分解剩余的硼氢化物,流水透析48 h,蒸馏水透析过夜,冷冻干燥得多糖醇。
将乙二醇处理后的溶液用蒸馏水透析48 h,袋外部分冷冻干燥做GC分析,袋内部分流水,蒸馏水各透析24 h,浓缩,加入NaBH4还原过夜。用50%醋酸中和至pH为6~7。浓缩至10 mL左右,取1/3干燥后做GC分析,剩余部分进行Smith降解。
1.2.4.6 加入等体积的0.2 mol/L TFA,使TFA终浓度为0.1 mol/L。35℃水解30 h,赶除TFA至pH为6~7,用蒸馏水透析48 h,袋外部分干燥做GC分析,袋内部分流水,蒸馏水各透析24 h,浓缩,做气相色谱分析,分析条件同1.2.1。
1.2.5 核磁共振(NMR)分析
将处理好的紫球藻多糖样品用D2O溶解,室温条件下,在Bruker AVANCE 400 Digital NMR型核磁共振仪上测试样品的氢谱和碳谱,对照文献资料分析图谱信息。
1.2.6 甲基化分析
1.2.6.1 紫球藻胞外多糖乙酰化衍生物的制备
0.5 g紫球藻胞外多糖加入到250 mL干燥的三口烧瓶中,加入20 mL甲酰胺,室温下搅拌30 min使紫球藻胞外多糖溶解,加入体积比为1∶1的吡啶-醋酸酐溶液20 mL,室温下反应24 h,然后加入250 mL蒸馏水反应除去多余的醋酸酐,混合液减压浓缩,浓缩液中加入无水乙醇醇沉,沉淀用无水乙醇洗涤三次,冷冻干燥,得到乙酰化多糖衍生物。
1.2.6.2 试剂的预处理:包括碘甲烷的纯化,二甲基亚砜的纯化和氢氧化钠粉的制备,方法参见文献[11]。
1.2.6.3 多糖的甲基化[12]
称取多糖8 mg于反应瓶中,真空干燥5~6 h,干燥后的样品加入用干燥4A分子筛处理过的3 mL DMSO,室温磁力搅拌直至多糖样品完全溶解后加入研磨过的干燥的NaOH粉末20 mg,室温搅拌10 min,然后改用冰浴5 min,待反应瓶中的溶液完全冰冻后,逐滴加入0.6 mL碘甲烷,同时反应物会慢慢地解冻,并逐渐澄清,直至成为亮黄色溶液,恢复至室温,继续磁力搅拌反应30 min,室温减压蒸馏,去尽过量的碘甲烷,然后用蒸馏水透析48 h,减压蒸至2 mL左右,进行冷冻干燥,然后真空干燥5 h,再重复上述操作5次后,取少量样品进行红外检测。
1.2.6.4 甲基化多糖水解和阿尔迪醇衍生物制备[12]
将已完全甲基化后的样品溶于3 mL 90%的甲酸溶液中,密塞,100℃下水浴解聚6 h,减压蒸干,加入2~3 mL甲醇,蒸干,重复三次以除尽过量的甲酸。然后向解聚后的样品中加入4 mL 2 mol/L TFA,密封后110℃下水解3~4 h,反应瓶中的溶液减压蒸干,再加入2~3 mL甲醇,蒸干,重复三次以除尽过量的TFA。水解后的样品用2 mL水溶解,加入20 mg NaBH4于室温下反应2 h,然后用冰醋酸调pH值至5左右,加1~2 mL甲醇及一滴冰醋酸再减压蒸干,重复三次,以除尽过量的醋酸。于110℃下干燥10~15 min后,加入3 mL的醋酸酐,100℃下反应1 h,减压蒸去未反应的醋酸酐,加入2 mL甲苯,减压蒸干,如此重复三次以除尽醋酸酐。最后将乙酰化后的样品溶于氯仿,再加入等体积的水洗涤氯仿层三次,除去水层,氯仿层加入无水硫酸钠干燥,放置10 min,过滤,将氯仿溶液浓缩至0.1 mL左右,作气质联用(GC-MS)分析。
1.2.6.5 GC-MS分析条件:升温方式为T=140 (3)-250/5。
2 结果与讨论
2.1 单糖组成分析
几种标准单糖按照保留时间从小到大分别是鼠李糖、岩藻糖、木糖、甘露糖、葡萄糖和半乳糖,它们在给定实验条件下的保留时间分别为4.67、4.73、4.85、6.56、6.64和6.70 min,内标物肌醇出峰时间为6.40 min。并且在4~7min范围内,各峰分离较好,基本无干扰现象。以标准单糖的保留时间为对照,采用糖醇衍生法对多糖样品进行前处理后进行GC分析,从图1中各个样品峰的保留时间可以推断1号、3号、4号峰分别为木糖(xyl)、葡萄糖(glc)和半乳糖(gal)的特征峰,它们的保留时间分别为4.83、6.62和6.69 min。内标法得到这三种单糖的摩尔比为2.96∶1.25∶3.06,三种单糖的含量百分组成分别为38.11%、17.91%和43.98%,半乳糖含量最高。该研究结果与 Geresh(2002)和 Gloaguen (2004)的报道基本一致[5,4],而与朱桂芳等[10]的研究结果紫球藻多糖由木糖、甘露糖、艾杜糖和葡萄糖组成有所不同。

图1 紫球藻多糖的单糖组成气相色谱图Fig.1 Gas chromatogram of monosaccharides composition of EPS

图2 紫球藻多糖的紫外扫描光谱Fig.2 Ultraviolet scan spectra of EPS
2.2 紫外光谱分析
紫球藻多糖的特征吸收波长位于220 nm,而在280 nm和260 nm波长处无吸收峰,表明样品不含游离或裸露的蛋白质和核酸(图2)。
2.3 红外光谱分析
图3显示的是紫球藻多糖的红外分析光谱。可见,在3600~3200 cm-1处(3419.56 cm-1)出现一个较强的宽峰,为羟基(-OH)的伸缩振动;在3000~2800 cm-1处(2927.43 cm-1)出现的吸收峰较弱,为C-H的伸缩振动,1400~1200 cm-1处(1384.79 cm-1)的峰是C-H的变角振动。这两组峰为多糖的特征吸收峰。1154~1022 cm-1范围(1120.20 cm-1)为C-O的伸缩振动,1259 cm-1位置的峰是硫酸基的非对称伸缩振动峰,931 cm-1附近的吸收峰是D-葡萄吡喃糖的环振动,该吸收峰较弱,说明为糖苷键类型为β构型[3]。

图3 紫球藻多糖EPS的红外光分析光谱Fig.3 Infrared spectrogram of EPS
2.4 碘反应
加入碘液后,糖液颜色没有变化,说明多糖结构中不含有连续的α(1→4)糖苷键结构,也证明不含淀粉或测试样品的空间结构与淀粉有较大区别[3]。
2.5 部分酸水解
多糖部分水解后,使多糖大分子裂解成较小的片断,适当控制实验条件,可以使多糖的骨架结构基本保持不变而使支链上或链末端的糖基水解下来。因此,进行部分酸水解(弱条件),可用于推断多糖的骨架结构及分支结构。
表1显示的是在三种水解条件下,袋外组分、上清组分与沉淀组分各单糖组成的数据。通过对这些数据的分析可以得到以下几个方面的推断:
2.5.1 在1.0 mol/L的三氟乙酸TFA作用下,沉淀量很少,说明水解进行的较完全。
2.5.2 Gal在0.05 mol/L的TFA弱酸水解条件下,袋外含量下降幅度较大,而上清组分中相对含量最高,表明在支链或链末端有较大量的分布。
2.5.3 Glc在TFA浓度由0.5 mol/L提高到1.0 mol/L,水解条件加剧时,上清中含量增加,提示其有可能分布在主链或靠近主链的区域。
2.5.4 Xyl、Glc在沉淀中的含量相对稳定,显示其在主链或靠近主链区域可能有特定分布。
2.5.5 0.05 mol/L和0.5 mol/L TFA条件下,上清中各糖含量大体稳定,说明被水解下来的支链和较长末端链的单糖分布可能比较均匀。
综上所述,可以推断 Xyl和Glc应主要分布在主链或靠近主链区域,Gal在支链或链末端有较大量的分布。
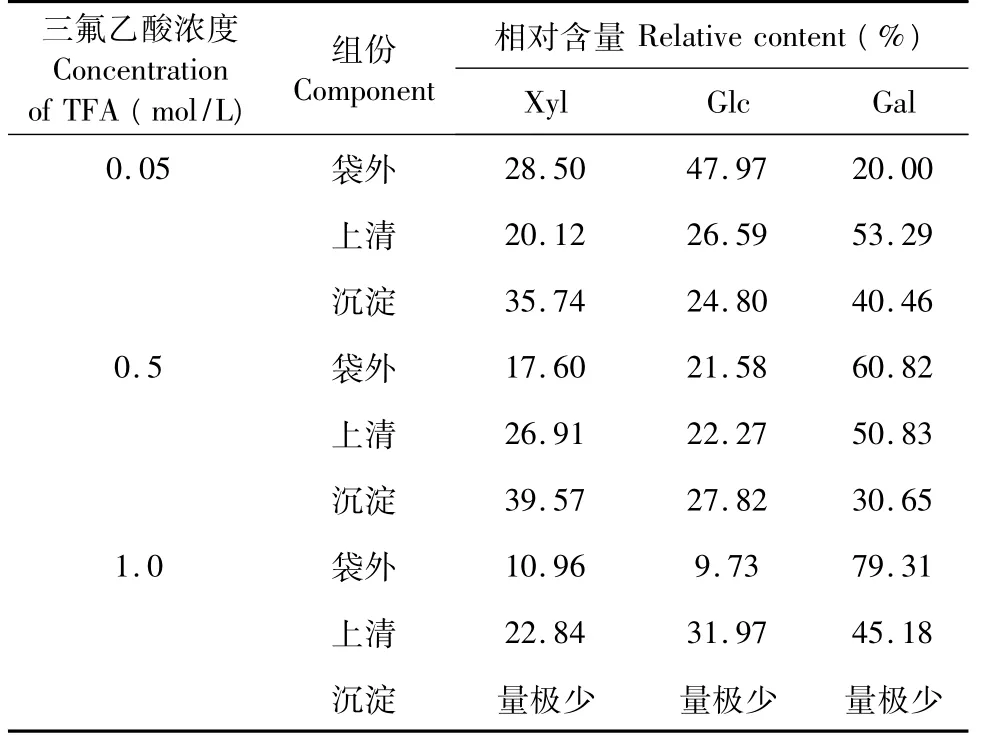
表1 部分酸水解后各单糖的相对含量Table 1 Relative content of monosaccharides after mild hydrolysis with TFA
2.6 Smith降解及高碘酸氧化
高碘酸氧化是一种选择性的氧化反应,它只能作用于多糖分子中连二羟基及连三羟基处。当连二羟基的C-C键被断开后,产生相应的醛;当连三羟基的C-C键断裂后,产生甲酸及相应的醛。此反应定量进行,每断开1 mol C-C键,消耗1 mol高碘酸,由此可知,每生成1 mol甲酸必然对应消耗2 mol高碘酸。因此,通过测定高碘酸消耗量及甲酸生成量,便可以判断糖苷键的位置、直链多糖的聚合度及支链多糖的分枝数目等[11]。
高碘酸氧化产物经NaBH4还原,得到的多糖醇用稀酸在温和条件下水解,可发生特异性降解,即Smith降解。Smith降解的特点是只打断被高碘酸破坏的糖苷键,而未被高碘酸氧化的糖残基仍连在糖链上。这样,多糖醇经Smith降解就可以得到小分子的多元醇和未被破坏的多糖或寡糖片段,对这些产物进行GC分析,便可以推断出糖苷键的键型及其位置[11]。
根据以上的原理,对紫球藻多糖进行了高碘酸氧化和Smith降解,并对其降解物做了全面的分析,得到的以下主要结果:
2.6.1 甲酸的测定结果,以酚酞做指示剂,氧化液滴加一滴NaOH溶液之后,即溶液即变浅粉色,说明甲酸含量极少,因此推测多糖分子中可能存在1→2、1→6糖苷键,但含量极少;而高碘酸消耗量大于甲酸生成量的两倍,说明可能存在1→2、1→2,6、1→4、1→4,6键型。
2.6.2 Smith降解之后,经过透析,对袋外组分进行GC分析,检测到甘油和赤藓醇。检出甘油,说明有产甘油键型,即1→2、1→6、1→2,6;检出赤藓醇,说明存在1→4或l→4,6等多种可氧化键型。但袋外组分未检测出单糖组分,可能是因为上述可氧化键型含量少或分布在主链或靠近主链的区域,经降解之后,只产生了少量的小分子片断。
2.6.3 Smith降解之后,袋内上清量多。经GC检测,袋内上清和沉淀两组分均检测出Xyl、Glc及Gal。结合②袋外组分GC分析结构,可以推断糖链中能被高碘酸氧化的键型少,而抗氧化的键型多,即含有大量的1→3位键(1→3,6;1→2,3;1→3,4;1→2,3,4类似)。
综合分析,紫球藻胞外多糖中糖苷键的连接方式主要为1→3,可能含有少量的1→6、l→2、l→4等键型。
2.7 核磁共振(NMR)分析
紫球藻多糖的1H NMR谱图如图4所示。其中4.7 ppm处是6-硫酸基-β-D-半乳糖C-1上H的化学位移;4.1 ppm处是6-硫酸基-β-D-半乳糖C-6上H的化学位移;3.6 ppm处是6-硫酸基-β-D-半乳糖C-2上H的化学位移。这三处化学位移表明样品EPS的硫酸酯基团连在C-6上。
紫球藻多糖的13C NMR谱图见图 5。其中68.53 ppm处为β-(1→3)-D-半乳糖C-2的化学位移。这处化学位移表明样品EPS含有以β-(1→3)糖苷键连接的D-半乳糖。由于紫球藻多糖样品分子量大,溶解性较差且粘度太大,致使1H NMR和13C NMR获得信息较少。但是样品的1H NMR中基本所有峰的化学位移均小于5.0 ppm,且较为集中,表明多糖的糖苷键为β型,与红外的结果一致。

图4 EPS的1H NMR谱图Fig.41H NMR of EPS

图5 EPS的13C NMR谱图Fig.513C NMR of EPS
2.8 甲基化和GC-MS分析结果
经乙酰化衍生化处理后,紫球藻多糖的水溶性大幅提高,达10 mg/mL,粘度明显降低。可见,采用乙酰化方法衍生紫球藻胞外多糖,可明显提高其水溶性,并可降低其粘度。
图6显示的是紫球藻多糖甲基化后,经水解、还原、乙酰化得到的甲基化糖醇乙酸酯衍生物的GCMS联机分析。可见,紫球藻多糖出现了5个甲基化衍生物离子峰,其保留时间分别为 7.620,14.268,18.703,19.974,26.691 min。经质谱数据分析它们分别为1,4-乙酰胺-3-氧-甲基-木糖,1-氧-乙酰基-2,3,6-三氧-甲基-D-葡萄糖,1,4,6-三氧-乙酰基-2,3-二氧-甲基-葡萄糖,(+)-甲基-半乳糖,D-半乳糖。
可以看出,紫球藻多糖EPS为一种主要含有1→3糖苷键,并含有1→4及1→6糖苷键的组成的杂多糖。
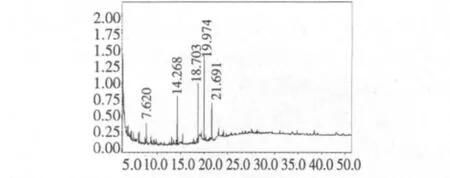
图6 甲基化糖醇乙酸酯衍生物GC-MS分析谱图Fig.6 GC-MS spectroscopy of methylated alditol acetated EPS
2.9 紫球藻多糖的重复单元结构分析结果
综合2.1至2.8的分析结果,可以推断紫球藻多糖EPS可能的重复单元为:
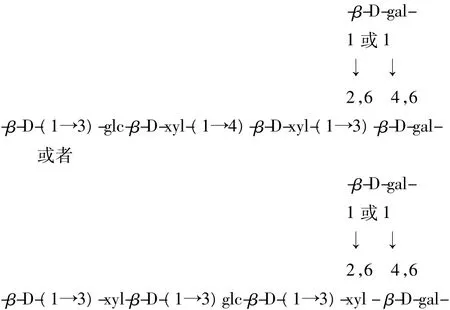
2.10 讨论
多糖的组成分析一般采用气相色谱法。由于样品在气相中传递速度快,可以选用广泛的固定相和灵敏的检测器,因而具有选择性强、分辨力好、灵敏度高以及分析速度快等优点[11]。采用气相色谱法测定糖类,遇到的主要困难是糖类本身没有足够的挥发性,所以必须在气相色谱分析之前预先转化成易挥发、对热较稳定的衍生物。目前常用的衍生物有糖的三甲基硅醚衍生物、糖肟三甲基硅醚衍生物、三氟乙酸酯衍生物、糖腈乙酸酯衍生物和糖醇乙酸酯衍生物等[11],由于糖的异构化,在某些衍生物的制备过程中,会产生衍生物的异构体,使得色谱分析使每一种糖产生几个峰,这往往影响组分的分离和定量。
在我们的前期研究中,对糖醇和糖腈乙酸酯两种衍生方法的气相分析效果进行了比较,结果发现糖醇乙酸酯衍生化方法能较好地实现其水解产物的衍生,与糖腈乙酸酯衍生法相比,避免了衍生物异构体的产生,每种糖都可以得到单峰且操作重复性强,适用于单糖的定量。此外,我们还注意到实验过程中多糖的水解是糖醇和糖腈乙酸酯两种衍物制备的关键,本研究采用2 mol/L三氟乙酸、120℃水解2 h的条件,可以将该多糖完全水解,避免了H2SO4水解多糖所带来的温度过高引起的爆管以及糖的炭化等问题,有效地缩短了水解时间。并且获得的衍生产物经减压浓缩蒸干,干燥,二氯乙烷溶解后作GC分析,得到的色谱峰未出现拖尾现象。
NMR用于多糖结构分析时,通常会因为多糖的高分子量而得不到更多的信号,本研究中为了获得更多地信息,将多糖样品进行进行了部分酸水解处理,但是仍然没有获得理想的结果;但是加大多糖的浓度(一般不低于5%)和升高温度可以提高分辨率,获得更多的信号峰。
多糖的甲基化分析是测定多糖分子中各单糖糖苷键连接位置的权威方法之一,但是多糖高的分子量会带来甲基化不完全的问题,影响最终结果的分析;此外,多糖大分子的空间折叠及分子内氢键的存在造成空间位阻使甲基化不完全[3]。本研究中,由于紫球藻多糖的分子量高达2.91×106Da,且水溶性较差,给甲基化实验带来很大的困难。将多糖乙酰化处理后,有效的改善了其溶解度,由于乙酰基在后续实验中会自动脱落,不会对实验造成影响。即便如此,由于试验条件的苛刻与本身实验条件的有限,再加上样品本身的大分子量都使甲基化进行的不完全,从而影响了实验结果的准确度,导致GCMS分析中无法给出更多的信息。因此,要想获得更多的紫球藻多糖的完整的糖链结构,还需克服上述实验中的一些不足,借助更多的仪器分析,如比旋光仪、多维NMR、扫描电镜法(SEM)、小角度X衍射法(SAXS)、多维MS方法等。
3 结论
3.1 GC分析结果表明紫球藻多糖主要由木糖、葡萄糖和半乳糖组成,它们的摩尔比为2.96∶1.25∶3.06,三种单糖的含量百分组成分别为38.11%、17.91%和43.98%,半乳糖含量最高。
3.2 IR分析表明紫球藻多糖具有多糖的特征吸收峰,为硫酸化多糖,且提示有β-D-葡萄吡喃糖存在; 1H NMR表明样品EPS的硫酸酯基团连在C-6上,半乳糖存在形式为6-硫酸基-β-D-半乳糖;13C NMR数据表明样品中D-半乳糖以β-(1→3)糖苷键连接。
3.3 碘反应显示糖链不含有连续的α(1→4)糖苷键;部分酸水解推断出Gal在支链或链末端有较大量的存在,Xyl和Glc在主链或靠近主链区域有特定分布;通过高碘酸氧化和Smith降解反应后单糖组成的变化,推断出糖链中存在大量的1→3型等抗高碘酸氧化的糖苷键,可能含有少量的1→6、l→2、l→4等键型;经甲基化后经GC-MS分析表明该衍生物为木糖、葡萄糖和半乳糖组成的杂多糖,连接方式主要有1→3糖苷键,并含有1→4及1→6糖苷键。
1 Zhuge J(诸葛健),Zhao ZF(赵振锋).Fang HY(方慧英).The relationship between structure and function of polysaccharides.J Wuxi Univ Light Ind(无锡轻工大学学报),2002,21:209-212.
2 Sheng SQ,Chermiak R.Structure of the capsular polysaccharide ofClostridium perfingensHobbs 10 determined by NMR spectroscopy.Carbohydr Res,1998,305:65-72.
3 Guo ZC(郭振楚).Sugar chemistry(糖类化学)Beijing: Chemistry Industry Press,2005.
4 Gloaguen V,Ruiz G,Morvant H,et al.The extracellular polysaccharide ofPorphyridiumsp.:an NMR study of lithium-resistant oligosaccharidic fragments.Carbohydr Res,2004,339: 97-103.
5 Geresh S,Adin I,Yarmolinsky E,et al.Characterization of the extracellular polysaccharide ofPorphyridiumsp.:molecular weight determination and rheological properties.Carbohydr Polym,2002,50:183-189.
6 Huheihel M,Ishanu V,Tal J,et al.Activity ofPorphyridiumsp.Polysaccharide against herpes simples viruses in vitro andin vivo J Biochem Bioph methods,2002,50:189-200.
7 Sun LQ,Wang CH,Shi QJ,et al.Preparation of different molecular weight polysaccharides fromPorphyridium Cruentumand their antioxidant activities.Int J Biol Macromol,2009, (45):42-47.
8 Irit D,Renven C,UrielSod M.et al.Solube polysaccharide and biomass of red microalgaPorphyridiumsp.alter intestinal morphology and reduce serum cholesterol in rats.British J Mutri,2000,184:469-472.
9 Gu NY(顾宁琰),Liu YF(刘宇峰).Antiirradiation activities of extracellular polysaccharide ofPorphyridiumsp.Mar Sci(海洋科学),2002,26(12):53-56.
10 Zhu GF(朱桂芳),Wei D(魏东),Liu SS(刘石生),et al.Seperation,Purif ication and Property of Extracellular Polysaccharide fromPorphyridiumsp.Nat Prod Res Dev(天然产物研究与开发),2005,17:542-548.
11 Zhang WJ(张惟杰).Study on Biochemistry Technology of Glycoconjugates(糖复合物生化研究技术),2st Ed.Hangzhou:Zhejiang University Press,2003,36-242.
12 Liu CH(刘纯慧),Ye L(叶亮),Lin QX(林亲雄),et al.Studies on preparation and antitumor activity of the polysaccharide from the eggs ofStrongylocentrotus nudus(SEP).Pharm Biotechnol(药物生物技术),2006,13:429-432
Analysis of Chemical Structures of Polysaccharide from Porphyridium Cruentum
SUN Li-qin1,2*,WANG Ling1,MA Cui-hua1,WANG Chang-hai2
1Life science college of Yantai University,Yantai 264005,China;2Bioscience and Engineering department of Dalian university of Technology,Dalian 116024,China
In this paper,the chemical analysis and spectrum analysis methods were used for investigating the level 1 sugar chain structure ofPorphyridium Cruentumpolysaccharide(EPS).The results of GC analysis showed that the sugar moiety of this polymer EPS was composed of three neutral monosaccharides(xyl,glc and gal),and molar ratio was 2.96∶1.25∶3.06.Infrared spectrum analysis showed that EPS was sulfuric acid ester polysaccharide,and its absolute configuration was β.The results of chemical analysis revealed that the main linkage of EPS was β-(1→3),and there were a few of 1→4 and 1→6 linkages.A large of galactoses exsited at the end or branch of sugar chain,but xylose and glucose were distributed specially close to main chain.NMR analysis displayed that sulfuric acid ester linked on C-6,and glycosidic bond was β-configuration.GC-MS on-line analysis further confirmed that EPS was a heteropolysaccharide,and sugar chains structure mainly contained 1→3 linkage and small number of 1→4,1→6 linkage.Based on above comprehensive analysis,the repeat units of polysaccharide produced by P.cruentum could be inferred as D-Xyl and D-Glc,which were present at the main chain with β-(1→3,1→4)linkage and D-Gal mainly existed at the end of main chain and branched chain.
Porphyridium cruentum;polysaccharide;sugar-chain structure;chemical analysis;spectrographic analysis
Q539
A
1001-6880(2012)05-0569-07
2011-07-05 接受日期:2011-12-06
国家“十一五”科技支撑计划项目(2008BAD94B04);山东省自然科学基金项目(Y2007D59,Y2008B21);烟台市科技发展计划项目(2009219)
*通讯作者 E-mail:sliqin2005@163.com
猜你喜欢
可再生能源(2022年8期)2022-08-17
天然产物研究与开发(2021年10期)2021-11-03
河北地质大学学报(2020年2期)2020-06-04
天然产物研究与开发(2019年1期)2019-03-01
大自然探索(2019年2期)2019-03-01
中成药(2018年8期)2018-08-29
中成药(2018年3期)2018-05-07
天然产物研究与开发(2016年1期)2016-06-05
中成药(2016年8期)2016-05-17
中华老年多器官疾病杂志(2016年7期)2016-04-28
