
HPLC法测定芯芭中木犀草素的含量
2012-09-12 11:43赵俊苹
中国民族医药杂志 2012年5期
赵俊苹 塔 娜 侯 敏
(1.呼和浩特市食品药品检验所,内蒙古 呼和浩特 010020;2.内蒙古食品药品检验所,内蒙古 呼和浩特 010020)
芯芭收载于《卫生部药品标准》蒙药分册[1]。根据“2009~2010年度国家药品标准提高工作科研立项综合意见单”增修订内容的要求,通过查阅有关文献[2],对芯芭中所含木犀草素进行了含量测定研究,参照《中国药典》2010年版一部[3]“北刘寄奴”项下的含量测定方法,以木犀草素作为指标成分,进行含量测定方法研究,经分析方法验证,该方法重现性好,专属性强,得到了比较满意的分析结果。
1 仪器与试药
1.1 仪器:岛津 LC-20泵,CBM-20A型控制器,SPDM20A型检测器,LC-Solution色谱工作站。
1.2 试剂与试药:木犀草素对照品(批号110823-200903,供含量测定用)中国药品生物制品检定所;芯芭由锡盟蒙药研究所提供。甲醇为色谱纯,水为高纯水,其它试剂均为分析纯。
2 方法与结果
2.1 色谱条件
2.1.1 色谱柱:色谱柱填充剂为十八烷基硅烷键合硅胶,本实验研究采用 ALLtima HP C18柱 (4.6×250mm)。流动相:乙睛 -0.3%磷酸(30∶70),流速:1.0mL/min;检测波长:347nm;理论板数按木犀草素峰计不得低于4000。
2.1.2 供试品溶液的制备:取本品粉末(过二号筛)约1g,精密称定,置具塞锥形瓶中,精密加入甲醇25mL,密塞,称定重量,超声处理(功率500W,频率40KHz)50min,放冷,再称定重量,用甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。
2.1.3 对照品溶液的制备:取木犀草素对照品适量,精密称定,加甲醇制成每1mL含8μg的溶液,即得。
2.1.4 阴性对照试验:按 2.1.2 与 2.1.3 项下的方法制备供试品溶液和对照品溶液。分别精密吸取甲醇空白溶液、对照品溶液、供试品溶液各10μL,分别注入液相色谱仪,测得结果为:空白溶液色谱图中在与木犀草素对照品以及供试品色谱图相对应的保留时间处无色谱峰出现,表明处方中其它组分对木犀草素的测定无干扰。见图(1、2、3)。
2.1.5 线性关系考察:精密称取木犀草素对照品约4.079mg,置100mL量瓶中,加甲醇使溶解,并稀释至刻度,摇匀。再精密吸取10mL,置50mL量瓶中,加甲醇稀释至刻度,摇匀,即得。(木犀草素 8.158μg·mL-1)分别取 1、2、5、10、15、20μL 进样,按上述色谱条件测定,以峰面积对进样量进行回归分析,结果木犀草素在8.158~163.16ng范围内呈良好的线性关系。回归方程为y=4306.6x-711.79;r=1。
2.1.6 稳定性试验 :取同一份供试品溶液,分别在0、2、6、10、12h进行测定,结果表明木犀草素在12h内的面积积分值基本稳定不变,RSD为0.47%。
2.1.7 重复性试验 :取同一批样品6份,各取粉末(过三号筛)约1g,测定每份样品含量,按外标法计算含量,结果平均含量为0.0205%,RSD为0.71%。
2.1.8 加样回收试验:取供试品(含量 0.205mg·g-1)6份,各约0.5g,精密称定,置6个具塞锥形瓶中,分别依次精密加入木犀草素对照品溶液(木犀草素0.0512)2.0mL,再分别精密加甲醇使成25mL,摇匀,按2.1.3项下方法操作,测定每份含量,计算回收率,结果见表1。
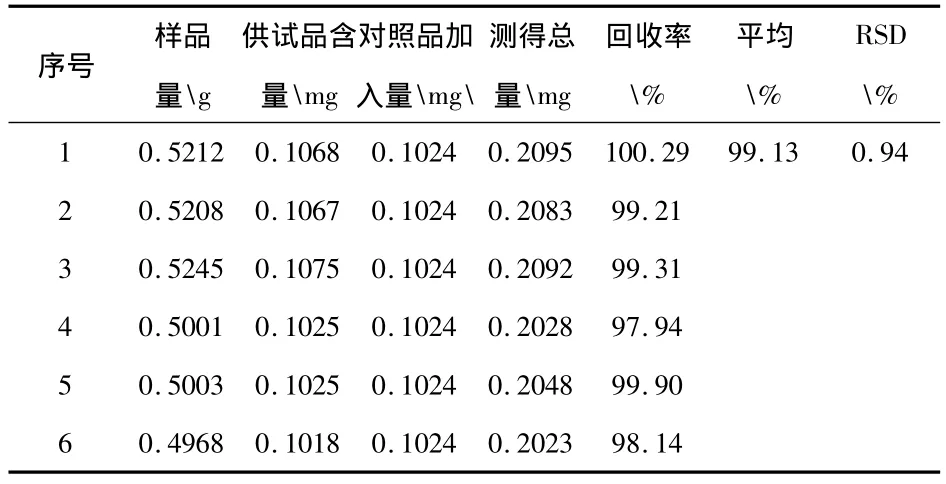
表1 木犀草素加样回收试验结果
2.1.9 样品含量测定:两批样品的测定结果见表2。
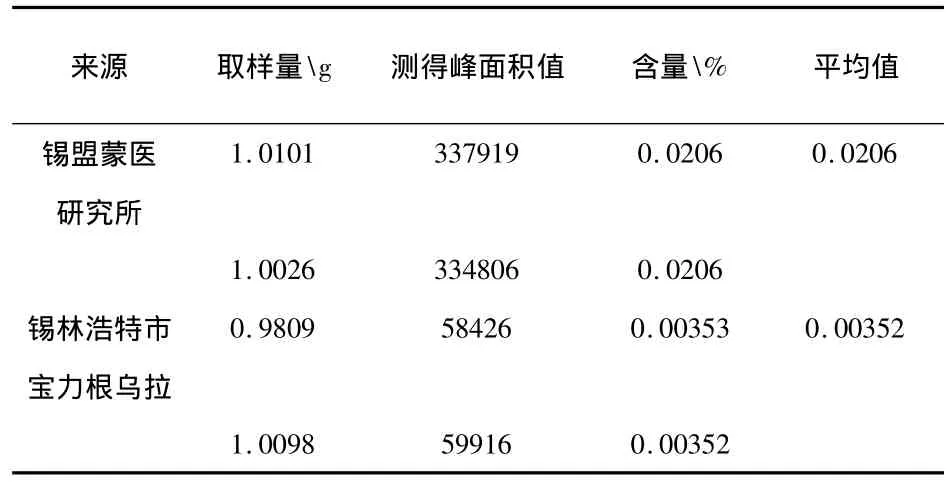
表2 样品中木犀草素含量测定结果

图1 阴性对照色谱图

图2 对照品色谱图
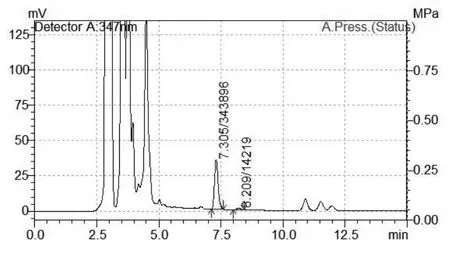
图3 样品色谱图
3 讨论
检测波长的选择:精密称取木犀草素对照品适量,用甲醇制成每1mL含25μg的溶液,通过岛津SPD-M20A型二极管阵列检测器对木犀草素自200~400nm波长范围扫描。结果木犀草素在347nm处有最大吸收。结合《中国药典》2010年版一部“北刘寄奴”药材项下含量测定方法,选择347nm作为检测波长。
[1]中华人民共和国卫生部药典委员会.卫生部药品标准蒙药分册[S].1998·18
[2]常新全,丁丽霞.中药活性成分分析手册[下册][M].学苑出版社,2002·1528
[3]国家药典委员会·中国药典[S].中国医药科技出版社,2010,91
猜你喜欢
北京联合大学学报(2022年1期)2022-02-12
天然产物研究与开发(2018年2期)2018-04-04
中成药(2017年12期)2018-01-19
西江月(2017年4期)2017-11-22
中成药(2017年4期)2017-05-17
中国药房(2017年6期)2017-03-29
小雪花·成长指南(2016年10期)2016-11-01
人民周刊(2016年11期)2016-06-30
中国医药生物技术(2014年4期)2014-01-23
郑州大学学报(理学版)(2012年4期)2012-03-25
