
一种新的图划分算法在PPI网络模块化中的研究
2012-09-11 04:30:30王晓芳贾宗维
山西农业大学学报(自然科学版) 2012年6期
王晓芳,贾宗维
(1.晋中师范高等专科学校,山西 晋中030600;2.山西农业大学 信息科学与工程学院,山西 太谷030801)
随着高通量生物蛋白质交互作用网络信息数据的不断扩大,越来越多的研究人员投身到预测蛋白质相互作用和蛋白质功能这一新兴的研究领域。众多研究表明,细胞中的蛋白质并不是孤立行动,而是通过彼此间的相互作用来实现特定的生物功能。蛋白质间的相互作用不仅关系到生物体的新陈代谢同时也是探究许多疾病病因的关键所在。因此,对蛋白质交互网络的研究对于蛋白质未知功能的预测以及了解细胞机制的结构有着十分重要的科学研究意义[1]。从高通量的生物蛋白质交互网络中通过聚类研究提取功能模块,有助于了解蛋白质相互作用机理和对未知蛋白质功能的预测。
近年来,酵母双杂交技术[2]、质谱分析技术[3]、蛋白质芯片技术[4]的发展和应用,使得高速增长的蛋白质相互作用数据可用于进一步分析研究。研究人员提出了许多蛋白质相互作用网络的聚类算法,用以分析蛋白质功能模块和预测未知蛋白质功能。它们大致分为基于层次聚类的方法、基于图划分和基于密度的局部搜索的聚类方法,如maximal clique方法[5]、RNSC方法[6]、MCODE方法[7]。本文在深入分析蛋白质网络的拓扑属性和蛋白质节点间的相互关系后,提出了一种基于蛋白质节点相似性的度量准则,以最优化模块度为目标函数的图聚类算法,用来发现蛋白质交互网络中的功能模块,实验结果表明所探测的功能模块内部的确是密集连接的。
1 数据及网络构建
1.1 数据来源
数据取自 Uetz等人[8]利用all-by-all筛选的技术在蛋白质组级别构建网络,“Ito-core”网络[9]及Yu等人[10]实验建立的一个高可信度的蛋白交互网络。以此3个实验数据集来验证算法所发现的蛋白功能模块划分的效果。
1.2 网络构建
用无向无权图 描述蛋白质相互作用网络,即G=(V,E),其中V表示图中节点的集合,E表示图中边的集合。蛋白质网络中每个蛋白质对应图中的一个节点,每条蛋白质间的相互作用对应图中的一条边。由此可以构造蛋白质网络关联图的邻接矩阵A,即
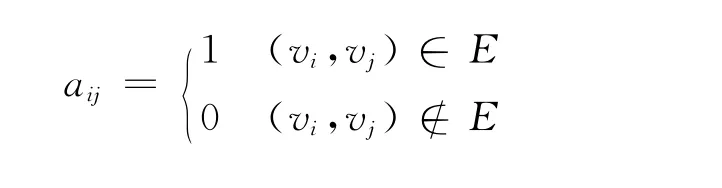
2 图划分聚类算法描述
蛋白质交互网络中的蛋白功能模块可以看作是构成完全图的各连通子图,探测功能模块的方法可以通过挖掘连通子图的图聚类方法。本文算法通过分析图中节点间的相互关系和节点间的最短路径数定义了节点间的相似性度量函数,利用模块度作为最优化聚类目标函数,逐层聚类直至最优解终止。
2.1 节点相似度定义
从图中的某一个节点出发去逐层遍访其余节点,同等时间内不同的节点访问的范围不尽相同。若两节点相似性较强,则遍访的范围基本一致,反之则差别太大。由此可见,两节点是否相似,与其周围的节点有很大关系。即两节点若关联的交集节点越多,则两节点就越相似。因此,可以用如下描述来定义两节点间的相似性度量准则。
假设蛋白质网络对应的图G是一个具有n各节点的无向无权图。则节点ti和tj间的相似性定义为:
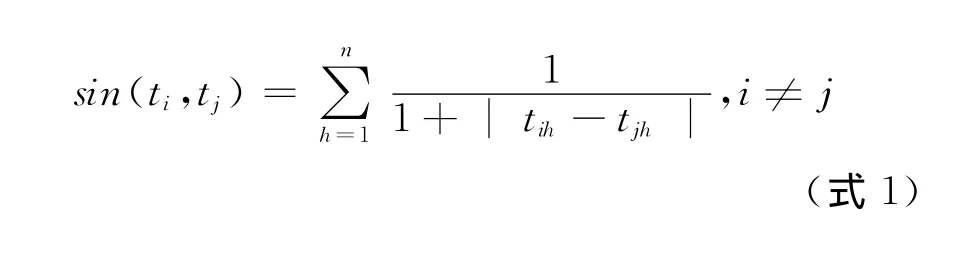
其中,tik表示从节点ti到达tk所要经过的最短步数(边数),由式1可获得图G中节点所对应的相似度矩阵S。
2.2 目标函数定义
模块度是用以衡量网络划分质量好坏的目标函数,当一个划分越接近真实划分时目标函数的值就越大,反之则越小。我们的算法就是通过对蛋白质交互网络进行层次聚类,同时计算目标函数值,选取目标函数值最大时的划分为最优解。目标函数为:
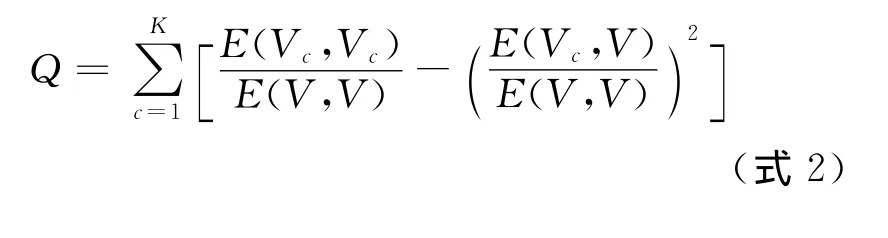
式中,K代表划分所得的功能模块数,E(Vc,Vc)表示模块C内部蛋白质相互作用边数,E(Vc,V)表示网络中与模块C中蛋白质有作用的边数,E(V,V)表示网络中的蛋白质相互作用总边数。
2.3 算法描述
将蛋白质网络数据预处理后,得到蛋白质的数目及蛋白质间的相互作用情况。算法描述如下:
1)通过蛋白质网络数据,构建蛋白质网络对应图邻接矩阵;
2)计算图中各节点对间的相似性值,建立相似度度量矩阵;
3)初始化网络为n个社团,即每个节点为一个社团;
4)合并图中目前相似性最大的节点对,并计算合并后的目标函数值;
5)重复执行步骤4,不断合并节点,直到整个网络合并为一个大社团。
算法中,目标函数的值并不随模块数的减少成反比,算法终止后,选取目标函数值最大时为最优,此时获得蛋白质网络模块功能划分为最优划分。
3 实验及分析
将算法应用于3个数据集构建的无向图。算法分别获得了0.68、0.75、0.64的目标函数最大值,模块划分效果较好,同时产生的若干功能模块则有待进一步的研究确认,这些高密度的功能模块能否正确的预测未知蛋白的功能还是未知数。但所发现的功能模块内部是密集连接的,说明模块内部很有可能存在许多蛋白复合物,许多信息都有待进一步挖掘。
对于实验组所发现的蛋白质功能模块,能否作为下一步研究中的蛋白功能预测候选模块,还需进一步验证各蛋白功能模块内部蛋白质相互作用密度是否符合要求。
定义蛋白相互作用密度函数来评价这些模块是否是内部密集连接。该指标描述如下:
子图B⊂G,B包含节点i。对于B而言,节点i度数分内外两部分,即ki=k(in)i(B)+k(out)i(B),即则
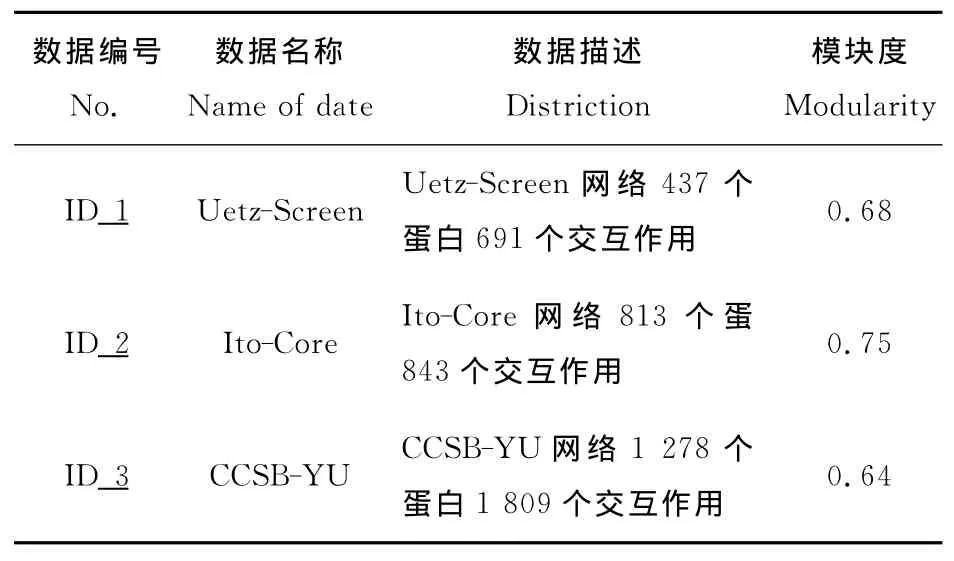
表1 各蛋白质网络数据实验结果Table 1 Experimental results of three data sets

通过对众多发现的子图密度计算得知,子图的连接密度大部分位于0.5~0.8之间,可见这些子图是完全可以作为下一步蛋白功能预测研究的可靠数据。
4 结语
采用节点相似度的图划分聚类算法,分析了3个蛋白质交互作用网络,对聚类得到的子图划分进行模块度函数的检测和单独模块的密度验证。研究表明,得到的功能模块划分的确是内部紧密连接的,能够作为蛋白质交互作用网络分析中功能预测的候选模块,从中挖掘相关的蛋白功能信息是下一步研究的重要任务。
[1]Yang Bo,Liu Dayou,Lin Jiming,et al.Complex network clustering algorithms[J].Journal of Software,2009,20(1):54-66.
[2]Fields S,Song O.A novel genetic system to detect protein-protein interactions[J].Nature,1989,340(6230):245-246.
[3]Ho Y,Gruhler A,Heilbut A,et al.Systematic identification of protein complexes in Saccharomyces cerevisiae by mass spectrometry[J].Nature,2002.415(6868):180-183.
[4]Zhu H,Bilgin M,Bangham R,et al.Global analysis of protein activities using proteome chips[J].Science,2001,293(5537):2101-2105.
[5]梅娟,王正祥,石贵阳,等.复杂生物网络分析的图聚类方法研究进展[J].食品与生物技术学报,2008,27(5):15-20.
[6]King AD,Przulj N,Jurisica I.Protein complex prediction via cost-based clustering[J].Bioinformatics,2004,20(17):3013-3020.
[7]Bader GD,Hogue CW.An automated method for finding molecular complexes in large protein interaction networks[J].BMC Bioinformatics,2003,4:2.
[8]Uetz P.A comprehensive analysis of protein-protein interactions in Saccharomyces cerevisiae[J].Nature,2000,403(6770):623-627.
[9]Ito T.A comprehensive two-hybrid analysis to explore the yeast protein interactome[J].Proc Natl Acad Sci,2001,98(8):4569-4574.
[10]Yu H.High-quality binary protein interaction map of the yeast interactome network[J].Science,2008,322(5898):104-110.
猜你喜欢
同济大学学报(自然科学版)(2019年2期)2019-04-02 05:43:48
同济大学学报(自然科学版)(2019年2期)2019-04-02 05:43:48
电子测试(2017年15期)2017-12-18 07:19:27
软件导刊(2016年9期)2016-11-07 21:35:42
电子科技大学学报(2016年2期)2016-08-31 02:50:00
通信电源技术(2016年5期)2016-03-22 01:09:49
石油知识(2016年2期)2016-02-28 16:20:16
智能系统学报(2015年4期)2015-12-27 09:38:39
自动化仪表(2015年11期)2015-04-01 01:02:40
电子设计工程(2015年6期)2015-02-27 12:04:53
