
水二聚体间相互作用的从头分子动力学研究
2012-09-07 08:29陈静
唐山学院学报 2012年3期
陈 静
(唐山学院基础教学部,河北唐山063000)
水二聚体间相互作用的从头分子动力学研究
陈 静
(唐山学院基础教学部,河北唐山063000)
应用Car-Parrinello从头分子动力学(CPMD)及限定性动力学方法对水二聚体在300K时不同距离和角度的相互作用能进行了研究,计算结果表明:水二聚体在R=3.,θ=170°附近形成的氢键结构最为稳定,其相互作用能量约为-18~-17kJ/mol。随着氧氧间距离R的增加,角度θ对氢键能量的影响逐渐减小,当R>4.时角度θ对氢键能量的影响已经很弱。
水二聚体;从头分子动力学;相互作用能
0 引言
水是很奇特的物质,它在许多的生物和化学过程中起着至关重要的作用。水的不寻常性质主要来源于水体系中的氢键网络。为了认识这种氢键网络,学者们对水体系中的氢键进行了广泛的研究,现在关于水体系的研究仍然是热门课题[1-7]。目前人们对于水体系中的氢键有了一定的认识,但认识还没有完全统一,例如:2004年Wernet等人[5]提出液态水体系中大多数水分子只形成两个强氢键,这个提法给了通常认为每个水分子形成四个氢键的观点以强烈的冲击和挑战;另一种不同的观点是水体系中氢键判别标准不是唯一的,还存在多样性[7]。这些争论表明我们对水体系中氢键的理解还远远不够,需要进一步地加深研究。
氢键的形成有来自供体-受体间的电荷转移的贡献,因此对氢键相互作用能进行计算时,采用从头量子力学方法更为适当[8]。Car-Parrinello提出的从头分子动力学方法(CPMD)[9],已经被用来对水体系及相关的现象进行了大量的深入研究[4,10-11]。水二聚体是研究液态水中氢键的重要模型,因为小的分子团簇更有利于从分子水平探究物质间的相互作用关系,找出其相互作用的物理本质,从而可以更好地理解液态水的宏观性质。氢键能量强烈依赖于距离和角度,所以本文应用CPMD及限定性动力学方法对水二聚体在300K时不同距离和角度的相互作用能进行研究,系统地认识常温下水分子相互作用能与水分子间取向的关系,并对水中氢键的判别标准给出合理的分析。
1 模拟方法与模拟细节
1.1 模拟方法
CPMD动力学方法是在赝势和平面波的基础上具体实现的[9-10],其动力学原理最重要的一点是在真实的物理系统中引入一个虚拟的电子动力学系统,这样得到一个虚拟系统的广义经典拉格朗日量:
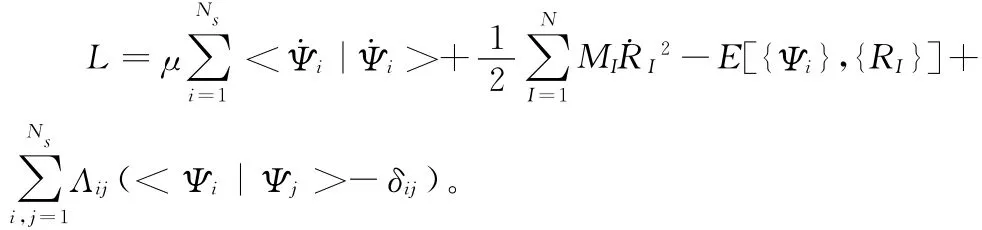
这里{Ψi}为单粒子轨道,μ为任意参数(单位为:Et2),从式子中看,μ相当于电子的“质量”,即虚拟质量,它起着调节电子运动时间标度的作用。MI和RI分别表示第I个原子的质量和位置。式中第一项和第二项分别是电子和离子的动能Ki和Kj,E[{Ψi},{RI}]是电子和离子耦合虚拟系统的势能。拉格朗日乘子Λij是为了保证波函数Ψi的正交性而引入的,在经典力学中,它是一个完整约束,在CPMD中将产生对运动的约束力。CPMD模拟过程中通过绝热退耦把核与轨道进行分离,这样可以使电子的运动轨迹始终趋向瞬时的能量最低点,即接近精确的Born-Oppenheimer表面。电子的虚拟质量μ与轨道是相关的,因此μ选择的参数必须合适,要求选取的参数尽量使核与轨道间的交换关联能可以忽略。
1.2 模拟细节
本文应用CPMD对孤立的水二聚体进行限定性动力学模拟,立方体盒子的边长为l=18.897 3a.u.,模拟采用Becke Lee-Yang-parr的交换关联泛函和范数不变的Troullier Martins赝势近似。Kohn-Sham轨道在平面波基组下展开,能量截断为70Ry,虚电子质量为600a.u.,时间步长设置为5a.u.(0.121fs),温度为300K,模拟过程中限定了二聚体O-O间的距离R和角度θ(参见图1)。

图1 水二聚体及相关的距离和角度
动力学模拟之前计算的孤立水分子的原子电荷分布、优化后水二聚体主要的几何参数以及优化后的水二聚体的分子轨道三个方面,与文献中报道的计算结果进行对比,以证实选用参数的可靠性。
限定性动力学模拟分为两个部分,第一部分模拟从优化结构开始,限定角度θ不变(θ=169.42°),改变O-O间的距离R,距离R每次变化0.01A,对于每一组R和θ均模拟20 000步;第二部分选取不同的O-O间的距离R,并限定R不变,改变角度θ,角度每次变化1°,对于每一组R和θ均模拟20 000步。根据Mark E.Tuckeman等人[10]对于氢键角度分布的分析结果我们确定了角度θ的范围为110°~180°。
2 结果分析与讨论
2.1 检验参数
为了验证所选用参数的合理性,我们从三个方面进行了检验:(i)计算孤立水分子的原子电荷分布;(ii)对水二聚体进行几何优化得到其优化后主要的几何参数;(iii)计算优化后的水二聚体的分子轨道。从表1,表2及图2中可以看出CPMD的计算结果与文献[12-14]给出的结果基本符合。以上三个方面可以肯定我们所选参数是合理的。

表1 孤立水分子原子电荷分布
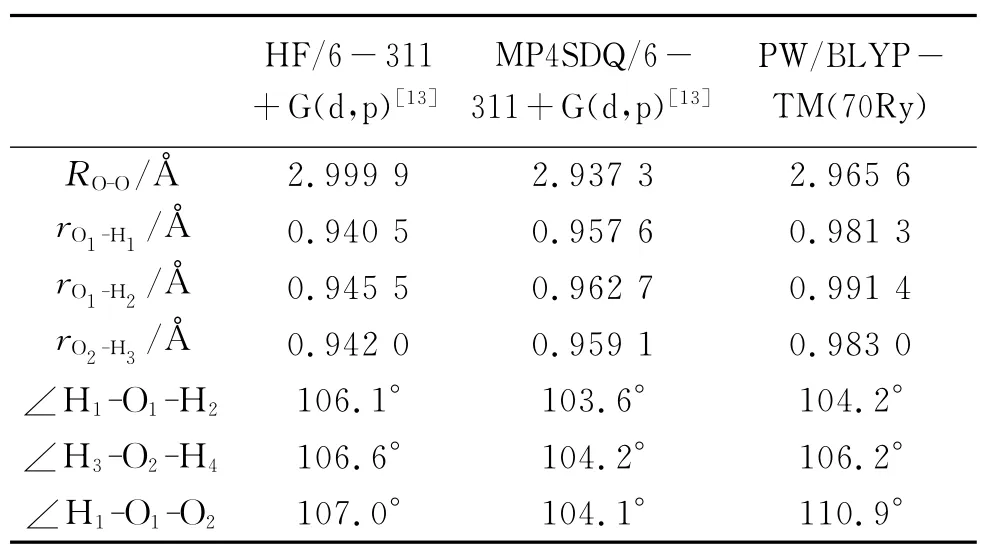
表2 水二聚体优化后的几何参数

图2 CPMD计算优化后水二聚体的分子轨道
2.2 水二聚体相互作用能与O-O间距离R及角度θ的关系
应该特别指出的是在研究水二聚体300K的相互作用能与分子取向的关系(结果如图3,4,5所示)时,动力学过程只限定了水二聚体的距离R和角度θ,其他的几何关系没有限定。在二聚体的模拟轨迹中可以观察到每一个水分子的键长、键角的振动以及水分子间的相对转动基本没有受到影响,二聚体在满足限定条件的热运动过程中遍历了几乎所有的可能构象,我们所给出的相互作用能是满足了距离R和角度θ的大量构象的平均值。

图3 相互作用能随距离R的变化
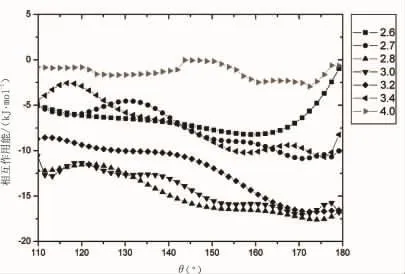
图4 相互作用能随角度θ的变化

图5 水二聚体300K时相互作用能的等高线
图3和图4所反映的300K时水二聚体相互作用能随距离和角度的分布特点使我们对于水中氢键能量有了直接的认识,可以看出R=3.,θ=170°附近是水二聚体最稳定的构型区域,且稳定区域对应的相互作用能大约为-18~-17kJ/mol,这与通常认为最稳定区域的氢键能量(大约-21 kJ/mol)不同,造成不同的主要原因是我们给出的相互作用能为热运动下大量构象的平均值,而不是某一个特定构象的相互作用能。热运动下的平均相互作用能可以更好地反映300K时水中氢键作用能的特点,这对于分析常温下液态水的宏观性质有着很大的帮助。从图4和图5中可以看出角度对于相互作用能的影响随着距离R的增加而减弱,当R大于4.时角度θ对于氢键能量的影响已经非常微弱,此时距离R在相互作用能中起着主要的作用。
目前氢键的判别标准可以分为能量标准、电荷转移标准、几何标准等。其中能量标准与电荷转移标准可以很好地反映氢键的物理本质,而几何标准在显示和统计方面有着很大的优势,因此被很多学者应用于氢键体系的研究。事实上任何一种判别标准都存在一定的人为性。如果我们按照氢键的能量标准(<-10kJ/mol[1,15])来判别是否成键,从图5可以看出依赖于距离R和角度θ的几何定义可以近似地满足方程θ≥(2 795-1 753R+287R2)°,其中θ<180°。
3 结论
应用Car-Parrinello从头分子动力学(CPMD)及限定性动力学方法对水二聚体在300K时不同距离和角度的相互作用能进行了系统的研究,计算结果表明:水二聚体最稳定构型区域对应于距离R为3.、θ角在170°附近,且稳定区域对应的平均相互作用能大约为-17~-18kJ/mol。
角度θ对于相互作用能的影响随着距离R的增加而减弱,当R大于4.时角度θ对于氢键能量的影响已经非常微弱,此时距离R在相互作用能中起着主要的作用。
从相互作用能等高线图中得到常温下水二聚体依赖于距离R和角度θ的几何定义满足方程θ≥(2 795-1 753R+287R2)°,其中θ<180°。
通过热运动下的水二聚体相互作用能研究可以更好地反映300K时水中氢键作用能的特点,这对于分析常温下液态水的宏观性质有着很大的帮助。
[1] Frank H Stillinger.Water Revisited[J].Science,1980,209(4455):451-457.
[2] Scheiner S.Ab initio studies of hydrogen bonds:the water dimer paradigm[J].Annu Rev Phys Chem,1994,45:23-56.
[3] Fecko C J,Eaves J D,Loparo J J,et al.Ultrafast hydrogen-bond dynamics in the infrared spectroscopy of water[J].Science,2003,301(5640):1698-1702.
[4] I-Feng W Kuo,Christopher J Mundy.An ab initio molecular dynamics study of the aqueous liquid-vapor interface[J].Science,2004,303(5658):658-660.
[5] Wernet Ph,Nordlund D,Bergmann U,et al.The structure of the first coordination shell in liquid water[J].Science,2004,304(5673):995-999.
[6] Jared D Smith,Christopher D Cappa,Kevin R Wilson,et al.Energetics of hydrogen bond network rearrangements in liquid water[J].Science,2004,306(5697):851-853.
[7] Kumar R,Schmidt J R,Skinner J L.Hydrogen bonding definitions and dynamics in liquid water[J].J Chem Phys,2007,126(20):204107-204118.
[8] Alexandre V Morozov,Tanja Kortemme,Kiril Tsemekhman,et al.Close agreement between the orientation dependence of hydrogen bonds observed in protein structures and quantum mechanical calculations[J].Proc Natl Acad Sci,2004,101(18):6946-6951.
[9] Car R,Parrinello M.Unified approach for molecualr dynamics and density-functional theory[J].Phys Rev Lett,1985,55:2471-2474.
[10] Hee-Seung Lee,Mark E Tuckerman.Structure of liquid water at ambient temperature from ab initio molecular dynamics performed in the complete basis set limit[J].J Chem Phys,2006,125:154507.
[11] Li Je-Luen,Car Roberto,Tang Chao,et al.Hydrophobic interaction and hydrogen-bond network for a methane pair in liquid water[J].Proc Natl Acad Sci,2004,104(8):2626-2630.
[12] Martin F,Zipse H.Charge distribution in the water molecule-A comparison of methods[J].J Comput Chem,2005,26:97-105.
[13] Hiroto Tachikawa.Electron capture dynamics of the water dimer:a direct ab initio dynamics study[J].Chem Phys Lett,2003,370:188-196.
[14] Pieniazek P A,Vande Vondele J,Jungwirth P,et al.Electronic structure of the water dimer cation[J].J Phys Chem A,2008,112:6159-6170.
[15] Jorgensen W L,Chandrasekhar J,Madura J D,et al.Comparison of simple potential functions for simulating liquid water[J].J Chem Phys,1983,79:926-935.
(责任编校:李秀荣)
Study on Interaction Energy for Water Dimer from Car Parrinello ab Initio Molecular Dynamics
CHEN Jing
(Department of Foundational Teaching,Tangshan College,Tangshan 063000,China)
The binding energy of the water dimer in different distance and angles at 300Kare studied from Car Parrinello ab initio molecular dynamics simulations combined with the constrained molecular dynamics method.According to the results of ab initio calculations,the global minimum energy is near to R=3.andθ=170°and the corresponding interaction energy of the stable region is about-17~-18kJ/mol.The effect of the hydrogen bond angle on the interaction energy weakens with the increase of the O-O separation,and when the distance Ris larger than 4.0,the effect of the hydrogen bond angle on the interaction energy is very faint.
water dimer;ab initio molecular dynamics;interaction energy
book=55,ebook=55
O561.4;O641.3
A
1672-349X(2012)03-0010-03
2012-03-29
2011年唐山市科学技术研究与发展指导计划项目(111302001a)
陈静(1978-),女,河北唐山人,讲师,硕士,主要从事分子动力学模拟研究。
猜你喜欢
波谱学杂志(2021年3期)2021-09-07
科教新报(2021年11期)2021-05-12
昆明医科大学学报(2021年1期)2021-02-07
心肺血管病杂志(2020年5期)2021-01-14
——以高中化学“氢键”的教学为例
教学月刊(中学版)(2020年13期)2020-12-29
意林原创版(2017年11期)2017-12-01
天津师范大学学报(自然科学版)(2016年4期)2016-12-14
中国卫生标准管理(2015年16期)2016-01-20
中学化学(2015年12期)2016-01-19
医学研究杂志(2015年6期)2015-07-01
