
TAK1siRNA对类风湿性关节炎患者滑膜细胞中mmp9和timp1表达的影响
2012-09-07 09:15兰忠煜白希壮韩晓锐
中国医科大学学报 2012年6期
兰忠煜,白希壮,韩晓锐
(中国医科大学附属第一医院骨外科,沈阳110001)
类风湿性关节炎(rheumatoid arthritis,RA)是一种慢性进行性自身免疫性疾病,以关节滑膜炎及对称性关节骨、软骨破坏为主要特征。其发病机制复杂,尚未完全清楚[1,2]。TAK1 属于 MAPKKK 家族成员。MAPKs途径通过MAPK激酶激酶(MAP3K),MAPK激酶(MAP2K,MEK)和MAPK依次被磷酸化的级联反应激活转录因子,调节特定的基因表达。P38通过靶向抑制炎性因子释放起作用[3],JNK又称c-jun氨基末端激酶是多数炎性疾病和代谢疾病的连接点[4],这些通路与RA联系非常紧密。关节软骨侵蚀是RA最为显著的表现,它的细胞外基质是由蛋白多糖类和胶原构成(主要是Ⅱ型胶原),这种结构的完整性是机械力学形成的关键[5]。滑膜细胞分泌的聚蛋白多糖酶(Aggrecanase)可降解蛋白多糖,基质金属蛋白酶9(mmp9)可降解Ⅱ型胶原。基质金属蛋白酶抑制剂1(timp1)能抑制绝大多数的mmp,可与mmp9前体及有活性的mmp9结合形成1∶1的化学复合物,此外还可以与明胶酶原B特异性结合使其失活。
1 材料与方法
1.1 细胞
11例滑膜组织标本来源于2009年11月至2010年12月中国医科大学附属第一医院骨科住院患者。所有患者诊断均符合美国风湿病学会1987年提出的诊断标准,术前均经伦理学认证。RA患者男4例,女7例,年龄50~64岁,平均56.2岁。
1.2 试剂
TAK1 siRNA及阴性对照由Thermo公司提供,靶向siRNA单链由4条构成,分别为GGACAUUGCUUCUACAAAU、GAGUGAAUCUGGACGUU
UA、GGAAAGCGUUUAUUGUAGA、GCAAUGAGU -UGGUGUUUAC。Real-timePCR试剂盒购自于TaKaRa公司,Vimentin试剂盒购自于ZYMED公司,相关引物均由金斯瑞公司合成,见表1。

表1 扩增TAK1、t i m p1、m m p9和G APD HeD N A基因片段的引物序列Tab.1 Pri m ersequences f oram pl i f i cat i on of cD N A f ragm ent s of TAKI,t i m p1,m m p9andG APD H Primer Sequence of primers Forward Primer 5′ATTCCACAGATACCAATGGCTC 3′TAK1 Reverse Primer 5′TGTAGTAACAATGCGATTTCGG 3′Forward Primer 5′TGACATCCGGTTCGTCTACA 3′timp1 Reverse Primer 5′TGCAGTTTTCCAGCAATGAG 3′Forward Primer 5′TGGGCAAGGGCGTCGTGGTT 3′mmp9 Reverse Primer 5′GGTCGTCGGTGTCGTAGTTGGC 3′GAPDH Forward Primer 5′AGGGCATCTTGGGCTACAC 3′Reverse Primer 5′TGGTCCAGGGTTTCTTACTCC 3′
1.3 实验方法
1.3.1 原代细胞提取及培养:DMEM和胎牛血清均购自Gibco公司。无菌切取滑膜组织,PBS反复冲洗,去除表面滑液。剪成糊状,置于1 g/LⅠ型胶原酶37℃消化4 h,再加入0.25%胰酶,37℃消化10 min,弃上清,200目纱网过滤,1 000 r/min离心10 min,弃上清,加入含10%胎牛血清的DMEM重悬细胞,接种于培养瓶内,置37℃、5%CO2细胞培养箱内培养。待细胞扩增至一定浓度,常规消化,传代培养。
1.3.2 细胞鉴定:将培养至3代的滑膜细胞做免疫组化鉴定,取出贴满细胞的盖玻片,10%甲醛溶液固定30 min。晾干后PBS冲洗3次,加入3%H2O25~10 min,PBS冲洗后,加入正常山羊血清、室温静置10~15 min,倾去、勿洗、甩干,加一抗过夜。滴加辣根过氧化物酶,室温孵育10 min,PBS冲洗3×3 min,DAB显色。显色完全后复染、脱水、封片。
1.3.3 细胞转染:取对数生长期的滑膜细胞种植于6孔板中,种板浓度3×105/孔。从6孔板中移除培养基,随机将培养的滑膜细胞分为阴性对照组,实验组。严格按照试剂说明书规定,每孔加入2 mL配制好的FECT(含特异性siRNA)转染液,培养细胞37℃、5%CO2孵箱中24 h,用于mRNA分析。
1.3.4 实时荧光定量PCR检测:提取RNA前0.5 h加入IL-1β置于孵箱中反应。吸尽6孔板中的转染液,加入1 mL Trizol,室温静置5 min。氯仿0.2 mL/mL Trizol,1 000 r/min离心15 min,将水相移入新管。异丙醇沉淀RNA,清洗完毕后加入反应体系反转。PCR反应总体积25 μL,其中SYBR Premix Ex.TaqTM2× 12.5 μL、上、下游引物各 0.5 μL,三蒸水 9.5 μL,模板 2 μL。循环参数:95℃ 5 min 变性,95 ℃ 1 min,60℃30 s,40个循环。最后延伸72℃5 min。
1.4 统计学处理
采用SPSS 16.0统计分析软件,计量资料均数比较用方差分析。P<0.05为差异有统计学意义。
2 结果
2.1 滑膜细胞的体外培养及鉴定
采用细胞培养法培养滑膜细胞,培养3 d后在其边缘有椭圆形、三角形或梭形成纤维样细胞长出,10 d左右细胞生长至培养瓶底面85%面积,消化传至3~5代的细胞生长迅速,细胞为长梭形、形态均一、排列规则。用Vimetin单抗标记后,经免疫组化发现:这些细胞均一地表达Vimetin(>95%)、表明所培养的细胞是滑膜成纤维细胞。见图1。
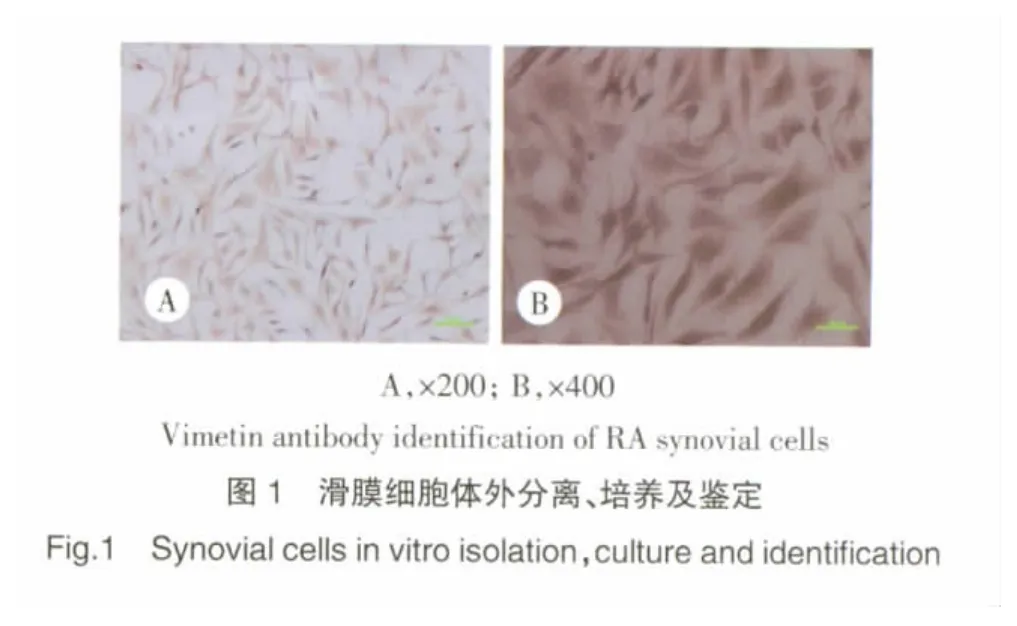
2.2 TAK1对timp1及mmp9表达的影响
从细胞中提取的总RNA,经分光光度计分析,其A260/A280值处于1.85~2.0之间。以上述总RNA为模板逆转录获得高纯度cDNA。以此为模板,荧光定量PCR扩增目的基因片段。用实时荧光定量PCR检测TAK1基因沉默效率大于80%。mmp9组实验组设定为1,对照组mRNA表达量3.967±0.551,明显高于实验组,差异有统计学意义(P<0.05)。沉默TTAK1前后,mmp9实验组降低率为74.76%。依上述统计方法timp1实验组设定为1,对照组mRNA表达量1.460±0.137,高于实验组,差异有统计学意义(P<0.05)。实验组沉默率31.51%(见图2、3)。设置空白对照组与阴性对照组比较无统计学意义。加入特异性TAK1siRNA后下调mmp9∶timp1比率。
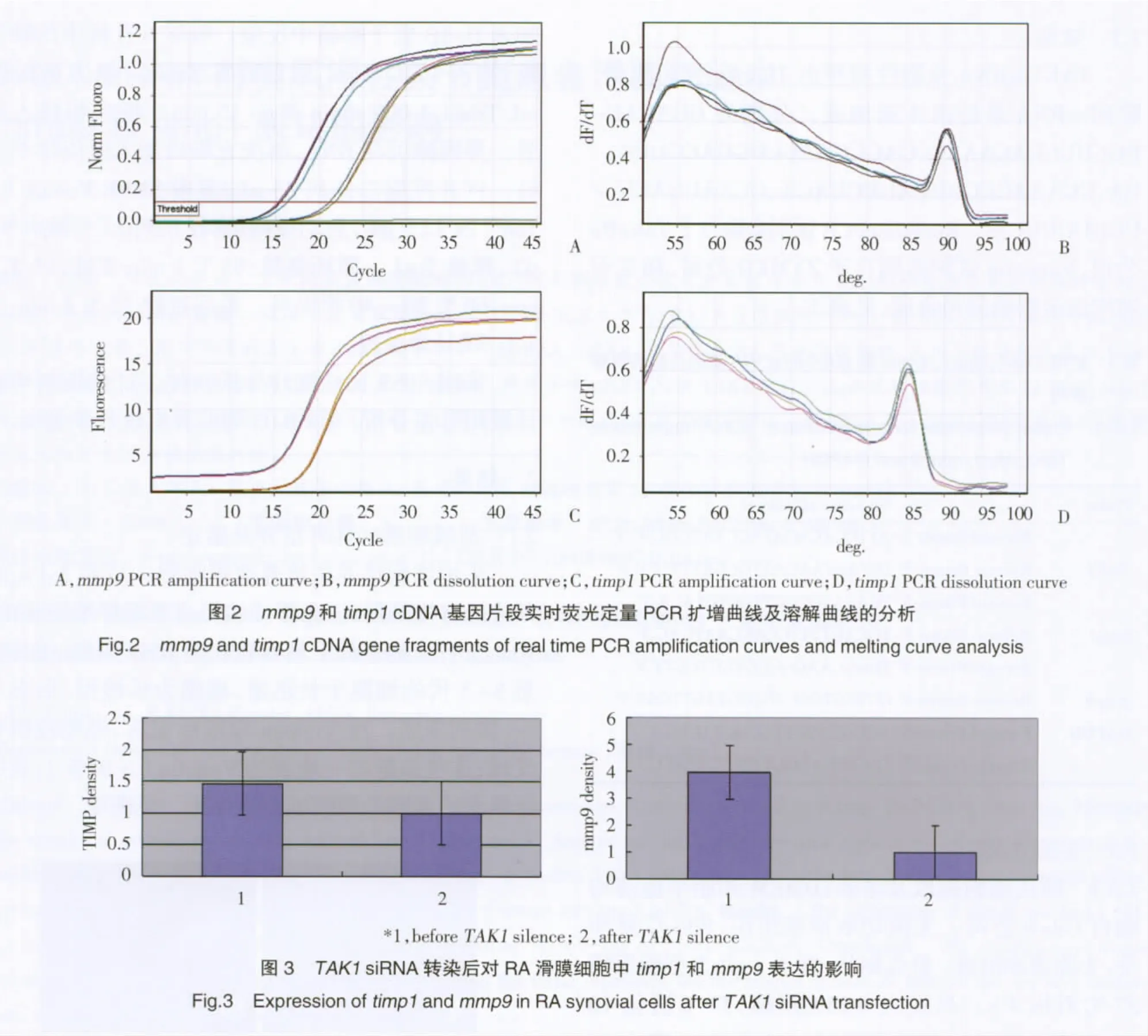
3 讨论
RA滑膜成纤维细胞(FLS)的活化是导致关节软骨破坏和关节炎性改变的主要原因。大量的炎性因子和炎性介质通过MAPKs途径诱导关节内炎性介质及MMPs的表达[6]。TAK1属于MAPKKK家族成员。MAPKs途径通过MAP3K,MAP2K和MAPK依次被磷酸化的级联反应激活转录因子,调节特定的基因表达。目前大量文献报道TAK1在类风湿关节炎滑膜组织中的表达量明显高于骨性关节炎和正常滑膜组织。我们的前期实验通过对RA滑膜、骨性关节炎滑膜和正常滑膜组织TAK1表达的研究也证实了这点。已经明确的FLS中调控炎症反应的通路大致有3条。MMPs超家族作为RA患者软骨破坏的主要成员,Hammaker[7]通过用 real-timePCR 实验在FLS中导入TAK1siRNA后mmp3表达水平明显下调。不过尚未见关于mmp9的相关报道。本实验为了避免体外实验细胞分化程度与体内有所不同,在行real-time PCR前30min于各组中加入IL-1β作为刺激因素。TIMP为MMP的天然抑制剂,timp1能抑制绝大多数的mmp,可与mmp9前体及有活性的mmp-1、3、9 形成 1:1 复合体,抑制其活性。Hegemann等[8]研究发现:timp1由巨噬细胞和结缔组织细胞合成并分泌,广泛存在于组织和体液中,并能被多种细胞因子所诱导,在巨噬细胞分泌的IL-1、TNF-α等细胞因子刺激下,并在被血浆激肽释放酶,纤溶酶或组织蛋白酶G等的作用下,转化为具有活性的mmp3的协同刺激下,并激活滑膜被覆细胞分泌的胶原蛋白酶,引发MMPs家族其他成员活化,进而使得滑膜被覆细胞和滑膜下层细胞同时合成timp1。MMPs和TIMP之间的比例关系决定软骨的破坏程度。
本研究发现运用转染技术高效抑制TAK1在RA滑膜细胞中的表达,real-time PCR发现timp1和mmp9的表达不同程度的降低,差异有统计学意义。特别是mmp9的表达量显著下调,这种作用在抑制RA患者软骨破坏中具有重要意义。我们在研究中发现沉默TAK1后mmp9mRNA降低率为74.76%。timp1sRNA降低率31.51%。两者之间的比例存在明显的不匹配关系。提示靶向抑制TAK1可打破MMP与TIMP之间的平衡关系,降低RA患者软骨的破坏。为治疗RA所导致的软骨破坏提供基因靶点。由于软骨破坏反应的调控是一个复杂的过程,涉及多个基因,多条途径,TIMP在滑膜细胞中的调控途径及调控机制尚未阐明,仍有待进一步研究。
[1]Stephen AP,Allan G,John FB.Rheumatoid arthritis1 In:Stephen AP.Manual of rheumatology and outpatient orthopedic disorders[M].天津:科技翻译出版公司,2003:192-291.
[2]任洁,李娟,冯知涛.类风湿关节炎患者滑液NK-22细胞对成纤维样滑膜细胞增殖的影响及其机制研究[J].南方医科大学学报,2011,31(4):661-664.
[3]CohenS,FleischmannR.Kinaseinhibitors:anewapproachto rheumatoid arthritis treatment[J].Curr Opin Rheumatol,2010,22(3):330-335.
[4]Liu G,Rondinone CM.JNK:bridging the insulin signaling and inflammatory pathway [J].Curr Opin Investig Drugs,2005,6(10):979-987.
[5]Dai SM,Shan ZZ.Implication of interleukin 18 in production of matrix metalloproteinases in articular chondrocytes in arthritis:direct effect on chondrocytes may not be pivotal[J].Ann Rheum Dis,2005,64(5):735-742.
[6]Geurts J,van den Brand BT.Toll-like receptor 4 signalling is specifically TGF-beta-activated kinase 1 independent in synovial fibroblasts[J].Rheumatology(Oxford),2011,50(7):1216-1225.
[7]Hammaker DR,Boyle DL,Inoue T,et al.Regulation of the JNK pathway by TGF-beta activated kinase 1 in rheumatoid arthritis synoviocytes[J].Arthritis Res Ther,2007,9(3):R57.
[8]Hegemann N,Wondium A,Ulricll K,et al.Synovial MMP-3 and TIMP-1 levels and their correlation with cytokine expression in canine rheumatoid arthritis[J].Vet Immunol Immunopathol,2003,91(3-4):199-204.
猜你喜欢
舰船科学技术(2022年10期)2022-06-17
昆明医科大学学报(2022年2期)2022-03-29
建材发展导向(2021年14期)2021-08-23
天津医科大学学报(2021年3期)2021-07-21
世界科学技术-中医药现代化(2021年12期)2021-04-19
中国临床医学影像杂志(2019年5期)2019-08-27
中国临床医学影像杂志(2019年1期)2019-04-25
中国医药生物技术(2015年4期)2015-12-26
中国民族民间医药·下半月(2014年2期)2014-09-26
中国医药导报(2011年27期)2011-12-31
