
金属卟啉仿生催化剂的研究进展
2012-09-01 06:07:38顾来沅
当代化工研究 2012年6期
顾来沅
(中北大学理学院,太原 030051)
金属卟啉仿生催化剂的研究进展
顾来沅
(中北大学理学院,太原 030051)
本文综述了近年来金属卟啉化合物的类型、制备方法及在催化剂方面的应用,以及仿生催化氧化机理的研究情况。迄今为止,已报道的金属卟啉催化剂大部分是小分子,在使用时难分离、不易回收且容易失活从而限制了其发展和应用。此外,有关其催化氧化机理的研究也报道甚少。该领域的发展趋势在于开发出可用于固载金属卟啉的新型材料和新的修饰组分,以及拓展金属卟啉在催化反应尤其是空气氧化体系中的应用。
金属卟啉;仿生催化剂;催化机理;新型材料
金属卟啉(MPs)是一类重要的仿生催化剂。它能够模拟细胞色素P450 单充氧酶, 在温和条件下活化分子氧, 使烃类物质在空气作用下高效率、高选择性、环境友好地得以催化氧化[1-2], 从而得到各种有机合成中间体, 满足工业生产的需求。1979 年 Grove[3]首次将人工合成的金属卟啉催化剂应用于有机底物的催化氧化,目前国内外对卟啉类化合物的研究非常活跃,特别是金属卟啉。金属卟啉在催化反应中存在氧化分解、自聚、失活, 以及难以回收等缺点,限制了在合成化学和工业领域的应用[4]。近年来, 将金属卟啉通过各种方法固载在各种载体表面, 形成化学结构和功能非常明确的固体催化中心, 从而把均相催化反应转化为多相催化反应,最终解决催化剂的回收问题, 目前已经取得了一系列研究成果。因此,本文综述了近年来金属卟啉仿生催化剂的研究进展,主要包括卟金属卟啉的类型、制备方法及其应用,以及仿生催化氧化机理,并展望了它的发展趋势。
1 卟啉化合物简介
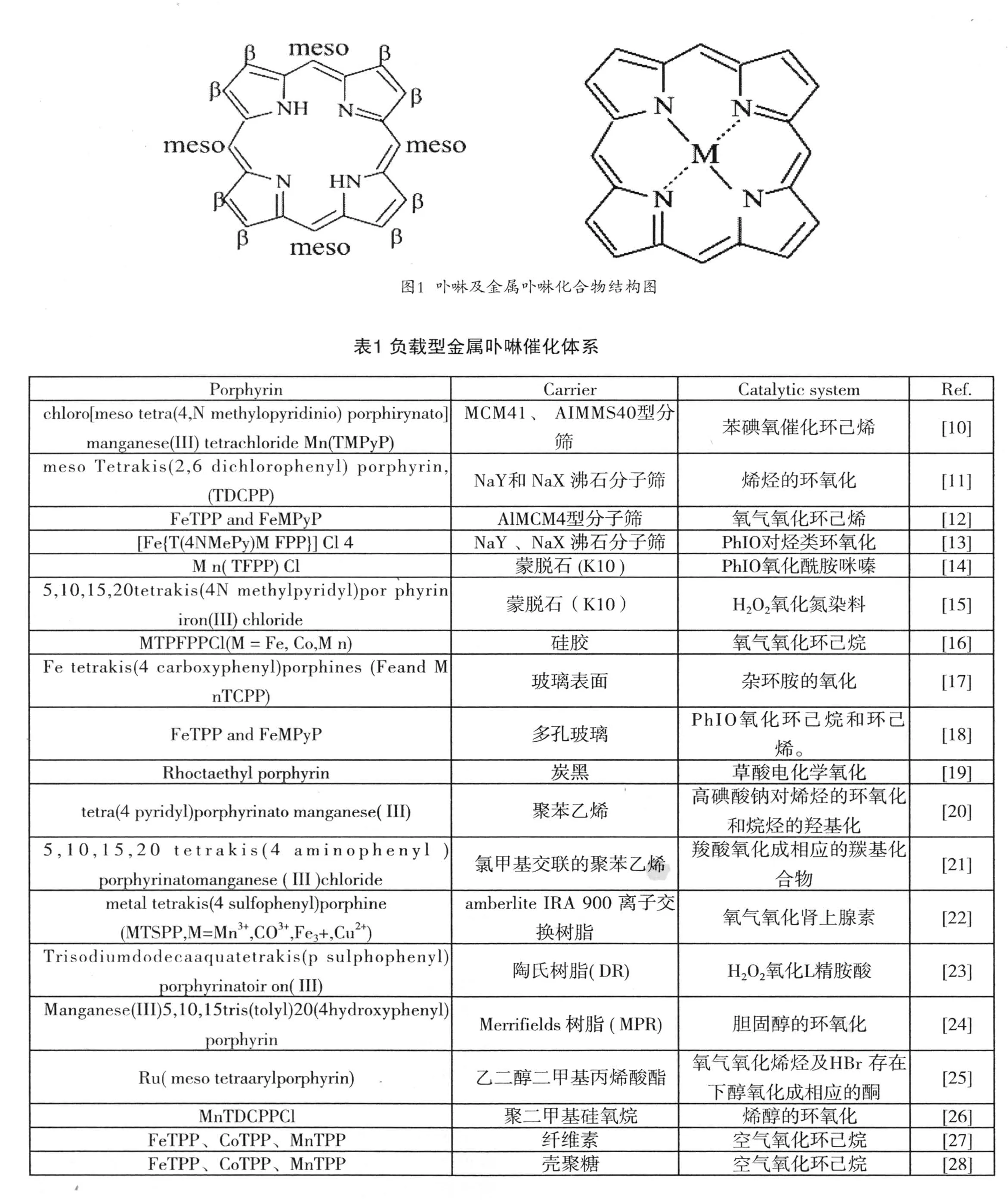
卟吩是由四个吡咯环通过次甲基键连形成的具有18个π 电子的大共扼体系。卟吩分子中4个吡咯环的8个β位和4 个中位(meso-)的氢原子均可被其他基团所取代,生成各种各样的卟吩衍生物,即卟啉,其中心的氮原子与金属原子配位形成金属卟啉配合物,金属卟啉的分子。
2 卟啉的合成方法
(l)Rothemund[5]方法:该方法在一段时间内一直是合成卟啉化合物的经典方法。它以醛类化合物(甲醛、乙醛、苯甲醛等)和吡咯为原料,以吡啶和甲醇为溶剂在封口的玻璃管中反应,水浴90℃~95℃下反应30min。该法所需反应条件苛刻,要求反应器密闭隔氧,底物浓度较低;而且后处理非常麻烦,反应收率低,仅有极少数芳醛可用于合成卟啉。
(2)Adler[6]方法:用吡咯和芳醛在丙酸介质中进行缩合反应,直接合成卟啉类试剂,提高了卟啉的产率。卟啉的产率主要受到溶液的酸碱性、溶剂的种类、反应温度和反应物的浓度等因素影响。但是这个方法有产率低、受反应条件的限制多、纯化困难等缺点。
(3)Lindsey[7]方法:采用苯甲醛和吡咯在氮气保护下,于二氯甲烷溶剂中用三氟化硼—乙醚作为催化剂,室温下反应生成四氢卟啉(TPC),然后以二氯二腈基苯醌(DDQ)氧化合成TPP,产率可达30%~40%。但是,这种合成方法反应浓度低,且最大反应容积为1L,放大后效果不好;需要无水无氧操作,且反应还不能一步生成TPP,必须在反应过程中另外加入氧化剂。
(4)微波激励法:利用微波辐射来加速有机反应、改变反应机理或开启新的反应通道。1992年法国化学家Petit[8]提供了一种新的合成方法:将吡咯和苯甲醛吸附于无机载体硅胶上,利用载体的酸性催化作用,在微波激励下合成四苯基卟啉,反应10min后直接用柱层析法分离,得到四苯基卟啉,收率为9.5%。
(5)郭灿城法[9]:采用DMF为溶剂,无水AlC13为催化剂,苯甲醛和吡咯缩合反应生成TPP,然后经过中性氧化铝柱分离,报道收率可达30%,高于Adler法,并且反应过程中无需氮气保护,产物中不含副产物TPC,反应时间也较短。该方法适用范围较广,对于以取代苯甲醛为原料的合成反应,产率为25%~30%;缺点是催化剂AICl3易与水反应生成Al(OH)3,给产物的分离造成困难。
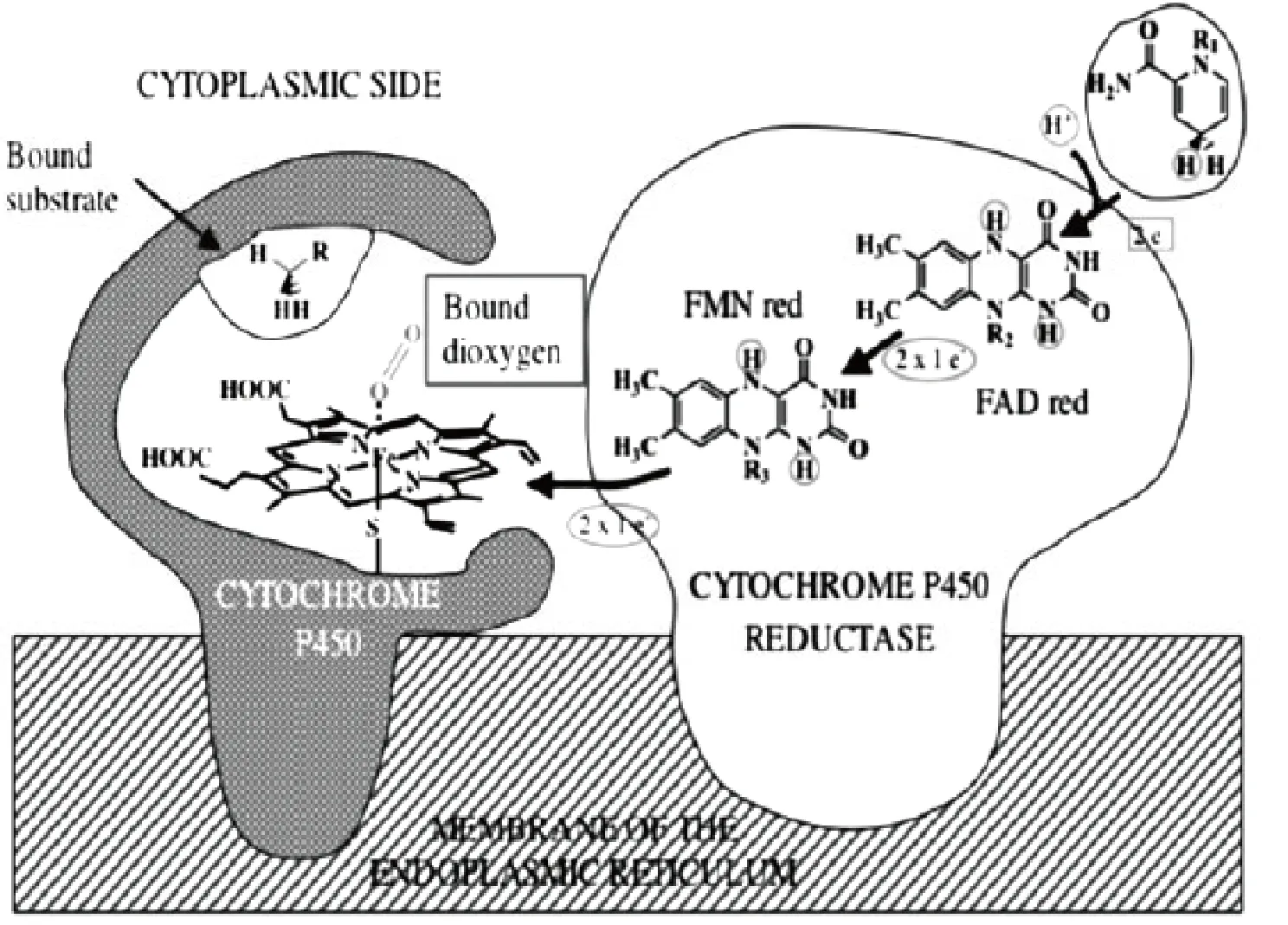
图2 细胞色素P2450及其催化过程
3 卟啉及金属卟啉化合物的分类
(l)按金属在元素周期表的位置,金属卟啉可分为主族金属卟啉、过渡金属卟啉和铜系、钢系金属卟啉。
(2)按金属原子在卟啉环上的结构,金属卟啉又可分为平面内型金属卟啉和平面外型金属卟啉。
(3)金属卟啉按其结构,又可以分为三类:a.不同金属原子鳌合的 TPP 类金属卟啉;b.在第一类金属卟啉大环体系的苯基上引入各类取代基后的衍生物,包括冠醚型卟啉、对面型卟啉等;c.在构成卟啉环的吡咯β位上引入氟,氯,嗅等原子。
(4)按固载的种类分:①通过静电吸引负载的金属卟啉; ②吸附在固体载体层或孔中的金属卟啉; ③通过形成轴向配体的固载金属卟啉;④以共价键的形式固载于聚合物表面的金属卟啉。本文列举了近年来国内外金属卟啉在固载催化领域应用进展如下:
4.金属卟啉化合物的仿生催化性能
4.1 细胞色素P450的催化机理
细胞色素P450是一类以铁卟啉为辅基的结合蛋白酶.它通过分子中铁化学价的变化在细胞的氧化还原反应中充当传递电子的使者,能催化许多有机物和分子氧之间的化学反应,是绝大多数仿酶催化的生物原型。近20年,很多学者对细胞色素P450在生物体内的催化氧化机理进行了大量的试验和理论研究。目前普遍接受的细胞色素P450的催化机理[29]如图2所示。
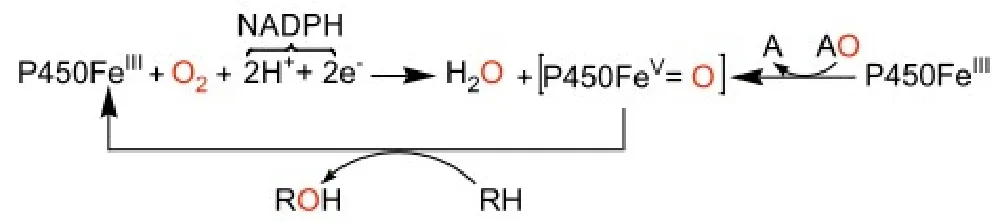
底物分子RH首先结合在细胞色素P2450酶的疏水区并把六配位的血红素中低自旋的Fe3+转变成五配位的高自旋Fe3+,配合物接受NADPH—烟酰胺腺嘌呤二核苷磷酸(是一种辅酶,叫还原型辅酶Ⅱ)传递的电子从而被还原至五配位的Fe2+配合物,接着再结合分子氧形成六配位氧合物。该氧合物继续得到1个电子使分子氧活化而形成高价Fe(V)=O活性中间体。该活性中间体将自身氧原子转移至底,释放产物ROH,并生成起始态的细胞色素P450,从而完成催化循环。该反应可以用公式概括为 :
4.2 金属卟啉仿生催化机理
对于金属卟啉催化剂催化氧化反应的机理报道很多,但都是自由基反应,起催化氧化作用的是高价铁氧自由基,且反应的机理只是假设的[30]。1999年, GYRGY[31]等报道了金属卟啉催化氧化甲基或亚甲基上的饱和C—H 键的羟基化,再次提出金属卟啉催化氧化反应是自由基反应,即TPPF202Fe4+O是催化氧化反应的活性物。LYONS等提出了一个与P2450在生物体内催化氧化机理相类似的铁卟啉仿生催化氧化假设机理[32],如图3所示。
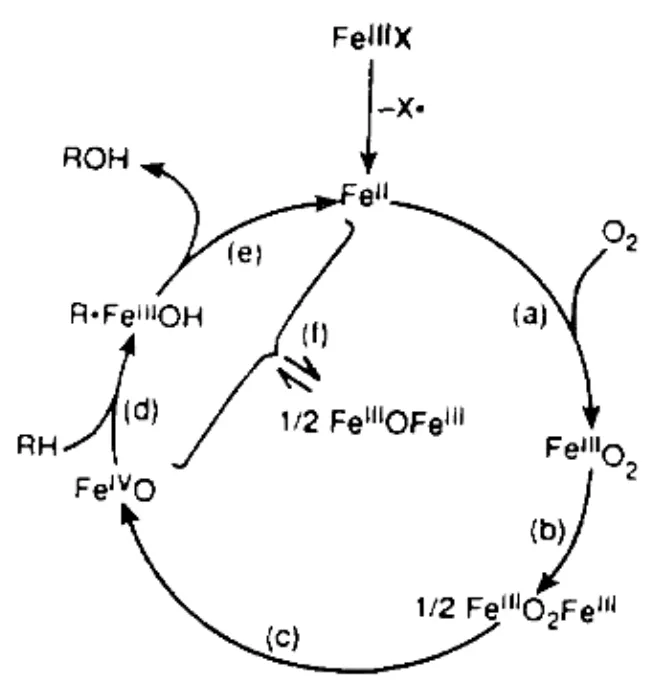
图3 假设机理
从图3可以看出其作用机理是:三价铁卟啉首先失去轴向配体自由基而还原为二价铁卟啉,二价铁卟啉与分子氧结合形成超氧铁卟啉络合物Fe3+O2,再与二价铁卟啉结合生成过氧铁卟啉络合物Fe3+-O2-Fe3+, Fe3+-O2-Fe3+断裂并生成稳定的高价铁氧络合物Fe4+O , Fe4+O氧化有机底物(如烃类RH)使之发生羟基化,与此同时,Fe4+O还原为三价铁羟基络合物Fe3+OH , Fe3+OH转化为游离三价铁卟啉 ,三价铁卟啉再从体系中得到一个电子变成二价铁卟啉 ,实现了一个完整的催化循环。
4.3 烃类仿生催化氧化过程与机理
金属卟啉催化空气氧化烷烃制备羟基化产物反应的特点是经历一个以催化剂活化分子氧为特征的烃类生物催化氧化循环过程。而工业上烃类氧化反应经历一个以断裂 C-H 键为特征的烃类自由基氧化的化学催化循环过程, 也就是烃类化合物在高温高压下均裂为烃基自由基, 烃基自由基与氧气结合产生烃基过氧自由基,烃基过氧自由基攫取烃的氢自由基产生过氧化物, 再生烃基自由基继续与氧气作用, 过氧化物在催化剂的催化下分解为烃的含氧化合物醇与酮。如图4所示。
由于把生物催化与化学催化相结合, 使得生物催化的烃基自由基能够进入到化学催化循环中, 避免了传统的需要高温高压才能把烃基 C-H键均裂为烃基自由基的过程。如下是THPPCoⅡ催化分子氧氧化环己烯的可能反应历程,如图5 所示。
从图5可见,金属卟啉催化分子氧氧化环己烯的反应主要发生在烯丙基碳原上,催化氧化主要产物为2-环己烯-1-醇(Ⅱ)和2-环己烯-1-酮(Ⅲ)。对此现象的解释目前有两种。Sawyer 等[33]认为,Fe(Ⅱ)( bpy)2配合物首先催化活化烯丙基的C—H 键,生成[( bpy)2 FeⅡ ( OOH)(c-C6H10)],再与分子氧反应生成高价态的[( bpy)2 FeⅣ(OOH) (c-C6H10)],与环己烯进一步反应生成环己烯醇、环己烯酮。Labinger[34]则认为,高卤代金属卟啉配合物催化分子氧氧化环己烯的反应是按照自由基机理进行的,反应发生在烯丙基碳上,金属卟啉配合物在反应中催化降解烯丙基过氧化氢,促进烷氧基自由基的生成。在 THPPCoⅡ的催化作用下反应得到 THPPCoⅣ—OH 和烷氧自由基,通过自由基链转移环己烯分子中烯丙基上的碳氢键在烷氧自由基作用下均裂为氢自由基和烷基自由基,而氢自由基和烷氧自由基发生链中止反应生成Ⅱ型产物,在分子氧的作用下可以进一步氧化为Ⅲ型产物。可见THPPCoⅡ在整个反应过程中对自由基的引发和传递有效地起着作用。
5 结语
卟啉类化合物在合成方面产率较低、条件苛刻以及过程比较复杂。因此,研究如何提高产率、如何优化反应条件,合成各个应用领域性能更好的卟啉试剂,具有非常重要的意义。在科学掌握了仿生催化氧化原理的基础上, 通过理论研究与技术开发可以实现仿生催化剂性能的改良优化, 从而可以达到改进生产工艺流程、节约自然资源、对环境友好, 以及极大地降低生产成本、提高生产效率的目的。
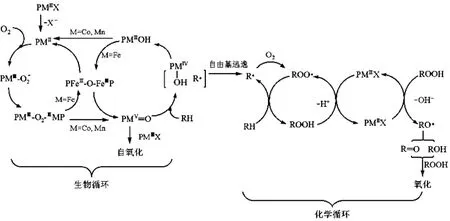
图4 金属卟啉催化循环和烃类自由基自氧化循环相耦联反应过程

图5 THPPCoⅡ 催化分子氧氧化环己烯的可能反应历程
[1]Yu X Q, H uang J S, Yu W Y, et al . J Am Chem Soc[ J], 2000, 122:5337~5342.
[2]Serwickaa E M , Poltowicza J, Bahranowskib K.Appl Cat A: General[ J],2004, 275: 9~14.
[3]Groves J T ,Nemo T E,Myers R S. Hydroxylation and epoxidation catalyzed by iron porphine complexes. Oxygentransfer from iodosylbenzene [ J ]. Journal of The American Chemical Society , 1979, 101 (4) : 1 032~1 033.
[4]M oghadama M , Shahroam T , Habibi M H . J of Molecu Cat A: Chem[J], 2004, 217: 912.
[5]ROTHEMUND P. Porphyrin Studies Ⅲ: The Structure of the Porphine Ring System [J ]. J Am Chem Soc ,1939 ,61 :2912~22915.
[6]Adler A D ,Longo F R ,Finarelli J D,et al. A simplified Synthesis for meso - Tetraphenylporphin[J ]. J Org Chem ,1976 ,32 :476.
[7]Lindsey S , Shreiman I C , Henry C Hsu , et al. Rothemund and Adler- Longo Reactions Revisited : Synthesis of Te 2traphenlyporphyrins under Equilibrium Conditions[J ].J Org Chem , 1987 ,52 :857~836.
[8]PETIT A ,LOUPY A. Microwave Irradation in Dry Media :A New and Easy Method for Synthesis of Tetrapyrrolic Com 2pounds [J ]. Synthetic Comm ,1992 ,22 (8) :1137~1142.
[9]郭灿城 ,何兴涛 ,邹纲要. 合成四苯基卟啉及其衍生物的新方法 [J].有机化学 ,1991 ,11 (4) :416~ 419.
[10]Serwicka E M , Poltow icz J, Bahranow ski K, et al .Appl Cat A:General[ J], 2004, 275: 914.
[11]Liu C J, Yu W Y, Li S G, et al . J Org Chem[ J],1998, 63: 7364~7369.
[12]Rani V R, Kishan M R, Kulkarni S J, et al. Cat Communi[ J], 2005, 6:531~538.
[13]Skrobot F C, Rosa L V, M arques P A, et al . J ofM olecu Cat A: Chem[ J], 2005, 237:86~92.
[14]Faria A L, M ac Leod T C O. Cat alToday, 2008,: 10. 1016.
[15]Barros V P, Faria A L, M acLeod T C O, et al .Biodeterioration &Biodegradat ion, 2007,: 10.1016.
[16]H aber J, M atachow ski L, Pamin K, et al . CatToday[ J], 2004, 92:195~198.
[17]A Iwado, M Mifune .A catalytic activity of glass beads, silica gels and anion-exchange resins modified with metal-porphines in oxidative reactions of ascorbic acid[J]Inorganica Chimica Acta 1998,283: 44~50.
[18]Shirley N, Immobilization of anionic iron(III) porphyrins onto in situ obtained zinc oxide Journal of Colloid and Interface Science, 2012,377:379~386.
[19]Poltow icz J, Serw icka E M , Bastardo Gonzalez E, etal . Appl Cat A:General[ J], 2001, 218: 211~217.
[20]梅文杰.新型固载金属卟啉:合成及对细胞色P450的化学模拟[J].化学学报2010, 218: 211~217.
[21]Dolphin D, Traylor T G, Xie L Y. Polyhaloporphyrins:unusual ligands formetals and metal- catalyzed oxidations[J]. Acc. Chem. Res., 1997,30: 251~259.
[22]计亮年, 刘敏. 各种金属卟啉催化环已烷的羟化作用[J]. 无机化学学报, 1991, 7(4): 420~ 423.
[23]Murahashi S, Naota T, Komiya K. Metallopor-phyrin- catalyzed oxidation of alkanes with moleccularoxygen in the presence of acetaldehyde [J]. TetrahedronLett., 1995, 36(44): 85 059~ 85 062.
[24]Dawson J H. Probing Structure- Function Relations inHeme-Containing Oxygenases and Peroxidasese [J]. Sci-ence, 1988, 240(41):433~ 439.
[25]LINDSEYJ S. Synthesis of Tetraporphrins Under Equilibrium Conditions [J].J Org Chem ,1987 ,52 (5) :827~836.
[26]PETIT A ,LOUPY A. Microwave Irradation in Dry Media :A New and Easy Method for Synthesis of Tetrapyrrolic Com 2pounds [J ]. Synthetic Comm ,1992 ,22 (8) :1137~1142.
[27]黄冠, 刘飞鸽,郭灿城. 高分子多糖载体对四苯基金属卟啉催化性能影响[J]. 化学学报, 2006, 7(64): 662~666.
[28]黄冠, 郭灿城. 壳聚糖固载的四苯基钴卟啉催化空气氧化环己烷[J]. 催化学报, 2005, 9(26): 765~768.
[29]Quentin Raffy ,et al. New biocatalysts mimicking oxidativehemoproteins: Hemoabzymes. C. R. Chimie ,2007 ,10 (8) :684。
[30]TAGLI A,TESTA P , PASTORINI A. Remarkable selectivity in the cyclopropanation reactions catalysed by an halogenated iron mesotetraphenylporphyrin [J].J Mol Catal A :Chem ,2003 ,198 :57~61.
[31]GY ,RGY M K,GY et al. Metallo-porphyrin catalysed biomimetic oxidation of aryl benzyl ethers :Implications for lignin peroxidase catalysis[J]. Tetra 2hedron ,1999 ,55 (14) :4 457~4466.
[32]LYONS J E ,ELLIS P E ,MA YERS H K. Halogenated metalloporphyrin complexes as catalysts for selective reactions ofa-cyclic alkanes with molecular oxygen[J]. J Catal ,1995 ,155 :59~73.
[33]J P Hage, J A Powell, D T Sawayer. J. Am. Chem. Soc.1995,117:12897~12898.
[34]J A Labinger. Catal. Lett. 1994,26:95~99.
Research Progress on Metalloporphyrins Biomimetic catalysts
Gu Laiyuan
(school of science, North University of China, Taiyuan 030051)
This review focuses the research progress on metalloporphyrins catalysts in recent decade ,including the type and the preparation and the application of metalloporphyrins catalysts in catalysis. It is pointed out that the majority of metalloporphyrins catalysts are micromolecule so far. The reported metalloporphyrins catalysts are very difficult to separate ,and not easy to recycle and easily inactivation,as a consequence,the development and application of metalloporphyrins catalysts are restricted.In addition,the catalytic oxidation mechanisms are reported little.it is necessary to develop new support materials or modifying agents to generate metalloporphyrins catalysts of novelty. Efforts should be devoted to find new application in catalysis,especially in reaction of air oxidation system.
metalloporphyrin; biomimetic catalysis; catalytic mechanism; new matarials
O631
A
T1672-8114(2012)06-09-06
顾来沅(1987—),女,硕士研究生,功能高分子材料。
猜你喜欢
化工生产与技术(2024年1期)2024-05-31 13:54:58
云南化工(2021年5期)2021-12-21 07:41:18
四川轻化工大学学报(自然科学版)(2021年3期)2021-08-30 06:36:44
中学生数理化·高二版(2017年3期)2017-07-07 12:17:51
中学生数理化·高二版(2016年2期)2016-05-30 07:50:39
合成化学(2015年10期)2016-01-17 08:56:06
山西大同大学学报(自然科学版)(2015年1期)2015-01-22 07:14:12
华东师范大学学报(自然科学版)(2014年4期)2014-03-11 16:18:28
中国造纸(2014年1期)2014-03-01 02:10:08
无机化学学报(2014年8期)2014-02-28 17:32:35
