
水汽液界面特性的平衡分子动力学模拟
2012-08-15 07:01王宝和王蕴博夏良志
河南化工 2012年21期
王宝和,王蕴博,李 群,夏良志,王 刚
(1.大连理工大学化工学院,辽宁大连 116024;2.大连理工大学化机学院,辽宁大连 116024)
水是许多化学反应过程廉价的反应溶剂,也是化工生产过程常用的工质。汽液界面行为是研究水相变传热问题的基础。目前,工程上许多有关水蒸发、水蒸气冷凝、加热干燥等相变传热数据仍主要依赖于实验。随着分子模拟技术的发展,采用分子动力学模拟方法,从分子水平揭示水汽液界面特性的研究,引起了国内外许多学者的极大关注[1-3]。本文拟采用SPC模型,对水汽液界面特性进行平衡分子动力学模拟研究,探讨温度、截断半径、模拟分子数对水汽液界面特性的影响规律。
1 模拟方法
1.1 模拟体系的建立
采用直角坐标系,模拟盒子如图1所示,液相位于模拟盒子的中央,汽相分别处于液相的上下两侧,整个模拟体系中有两个汽液界面。模拟盒子在x、y方向的长度为Lx=Ly=L,在z方向的长度为Lz=3L。

图1 模拟盒子的示意图
对于水的分子动力学模拟研究,采用的势能模型有很多,如 SPC、SPC/E、TIP3P、TIP4P、TIP5P等[4-5]。本文采用SPC刚体势能模型,假设只有不同水分子的O原子之间存在短程L-J势能,不同水分子的H原子之间以及H原子和O原子之间存在长程静电势能。水分子的总势能由短程L-J势能和长程静电势能两部分组成,如式(1)所示[2,3]。SPC模型的势能参数如表1所示,其中qH和qO分别为水分子中H原子和O原子所带电荷,rOH为H原子与O原子之间的键长,θ为两个O—H键之间的角度(即键角),σO为O原子之间L-J势能的尺度参数,εO为O原子之间L-J势能的能量参数,e为基本电荷(1e=1.6×10-19C),kB为 Boltzmann常数(kB=1.3806 ×10-23J/K)。

表1 SPC模型的参数值
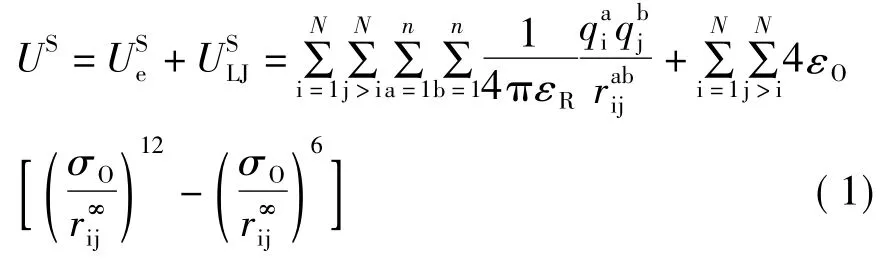
式中:US为总势能,kJ/mol;为长程静电势能,kJ/mol;为短程L-J势能,kJ/mol;N为模拟分子个数;n为每个水分子内受静电作用的作用点数量;i、j为模拟系统内2个不同的水分子;a、b为分子受静电作用的作用点;为i分子中a作用点所带电量,C;为j分子中b作用点所带电量,C;为i分子中a作用点与j分子中b作用点之间的距离,m;εR为真空中介电常数,εR=8.854 ×10-12F/m;i分子和j分子两个O原子之间的距离,m;σO为O原子之间L-J势能的尺度参数,m;εO为O原子之间L-J势能的能量参数,kJ/mol。
对于长程静电势能,采用作用场法。为避免L-J势能和静电势能在边界处发生截断而不连续,导致Hamiltonian函数不守恒问题。采用移位法来处理两种势能,如方程(2)和(3)所示[2-3,6]。
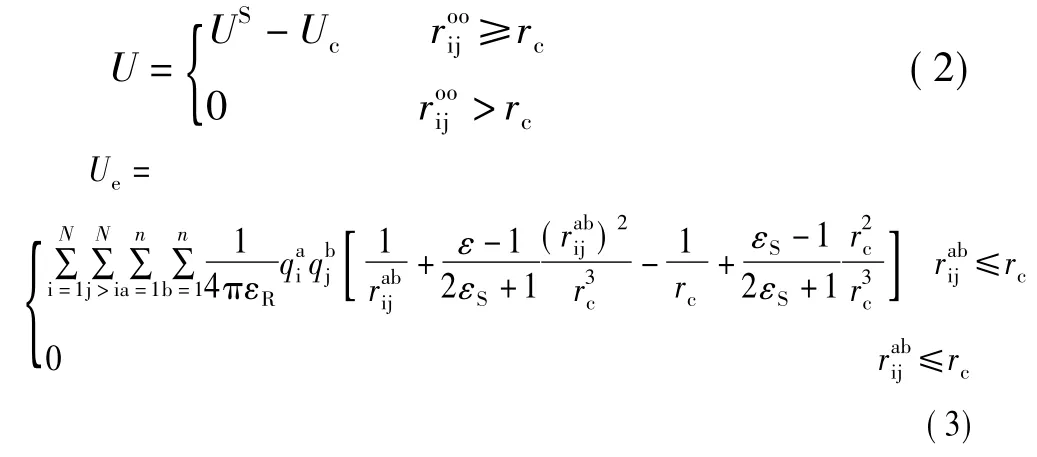
式中:rc为截断半径,m;U为校正后的势能,kJ/mol;Uc为截断半径处的势能,kJ/mol;εS为环境介电常数,通常取 εS= ∞[2,3,6],因此,式(3)可以简化为方程(4)。
1.2 模拟细节
初始时刻,水分子初始位置为各分子的质心以面心立方晶格(FCC)均匀排列在边长为L的液相模拟盒中,液相区上下两侧的汽相区为真空。水分子质心(即O原子所在位置)为分子坐标的原点,H和O原子均在xy平面上,其中一个H原子位于x轴的正方向上,另一个H原子位于xy平面的第二象限区,O和H的位置矢量分别为rO(0,0,0),rH(0.3159 σO,0,0),rH( -0.1053σO,0.2978σO,0)。水分子初始平动速度由随机数发生器随机给定,初始转动速度为0。
在模拟过程中,对物理量进行无量纲化处理;x、y、z三个方向均采用周期性边界条件;保证系统的体积V、温度T和模拟分子数 N保持不变,采用Woodcock变标度恒温法实现系统恒温;不断对体系质心进行矫正,使之处于坐标原点;将模拟盒子沿z方向划分为300个等厚度的薄片;模拟时间步长为0.8fs,总模拟步数为60万步,其中前20万步用于使系统达到平衡,后40万步用于统计界面特性参数。
模拟计算程序是由本课题组采用Fortran语言编写的,其模拟流程如图2所示。模拟运算中所涉及到的方程如式(5) ~ (13)所示[2-3,6]。



图2 模拟流程简图
式中:U(k)为第k个切片的势能,Uij(k)为i、j分子在第k个切片内的势能,nk为第k个切片的分子数,Vs1为切片的体积,ρ(k)为第k个切片的数密度,rij为 i分子和 j分子之间的距离,xij、yij、zij为 rij分别在 x、y、z方向上的分量,、、分别为 i分子中的a原子和j分子中的b原子之间的距离在x、y、z方向上的分量,U()为势函数 U()对的导数,PN(k)、PT(k)分别为第k个切片的法向应力和切向应力,γ(k)为第k个切片的局部界面张力,Δz为切片厚度,γ为汽液界面张力,〈〉为系统统计平均,ρV、ρL分别为汽相主体、液相主体密度,NL、NV分别为液相、汽相切片数,UV、UL分别为汽相主体、液相主体势能(L-J势能、静电势能、总势能),z(k)为第k个切片的位置,z0为Gibbs汽液界面的位置,d为汽液界面厚度。在统计切片内法向应力和切向应力时,若相互作用的原子a,b均在同一切片内,则计算全部作用;若相互作用原子只有一个原子在某一切片内,则计算一半作用。
2 模拟结果与讨论
2.1 温度对密度分布的影响
在模拟分子数N=256和截断半径rc=0.9498 nm 的条件下,当温度 T=400、450、500、550 和610 K时,模拟得到的密度分布如图3所示。统计得到的汽相主体密度ρV、液相主体密度ρL及汽液界面厚度d如表2所示。由图3及表2可见,汽相主体密度和汽液界面厚度随温度的提高而增加,而液相主体密度随温度的提高而减小。

图3 温度对水密度分布的影响

表2 不同温度下水的汽相主体密度、液相主体密度及界面厚度的模拟结果
液相主体密度与汽相主体密度之差(ρL-ρV)与温度T的关系如图4所示。可见,液、汽相主体密度之差随温度的升高而降低;从理论上讲,在临界点处,其差值应该趋近于零,这与图3所示的规律一致。液、汽相主体密度之差与温度的关系可以拟合成式(14)的形式。

式中水临界温度Tc=647.3 K,利用表2数据对式(14)进行拟合,得到参数 ρ0=1545.8 kg/m3,指数因子x=0.5516。

图4 液、汽相主体密度之差与温度之间的关系
2.2 温度对界面张力的影响
在模拟分子数N=256和截断半径rc=0.9498 nm 的条件下,当温度 T=400、450、500、550 和610 K时,水汽液界面张力的模拟结果见表3。

表3 不同温度下汽液界面张力模拟结果
图5为局部界面张力的分布曲线(500 K)。由图5可见,汽相主体的局部界面张力基本为零;从汽相主体向液相主体的过渡过程中,界面张力值逐渐增加,在汽液界面区达到峰值;在液相主体又在零值附近波动。水汽液界面张力模拟值随温度变化规律如图6所示。

图5 局部界面张力的模拟分布曲线(500 K)

图6 界面张力与温度的关系曲线
由图6可以看出,随着温度的提高,界面张力降低,模拟值与实验值之间的误差逐渐减小。界面张力与温度的关系可以拟合得到方程(15)。

将表3的数据对式(15)进行拟合,得到的参数γ0=254.3 mN·m-1,指数因子 k=1.305。
2.3 温度对势能分布的影响
在模拟分子数N=256和截断半径rc=0.9498 nm 的条件下,当温度 T=400、450、500、550 和610 K时,汽相主体总势能UV、液相主体总势能UL及总势能势阱深度ΔU的模拟结果如表4所示。图7为水分子的势能分布曲线(500 K),图8为液相主体区域的势能随温度的变化趋势。
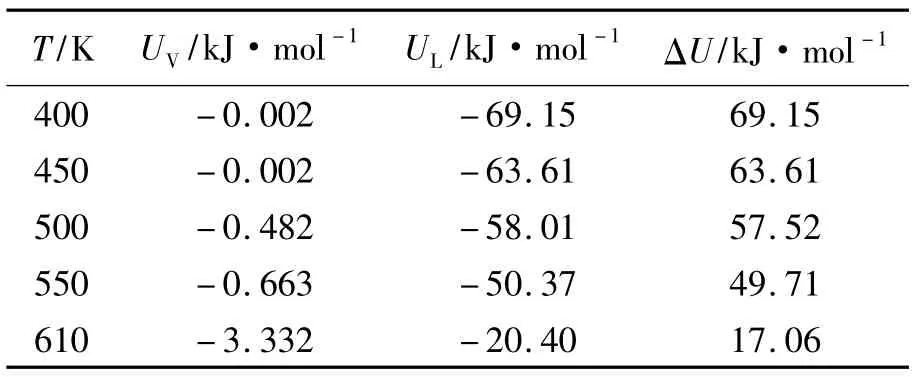
表4 不同温度下水分子势能的模拟结果

图7 水分子的势能分布曲线(500 K)

图8 液相主体区域的势能随温度的变化趋势
前已述及,水的势能分为L-J势能和静电势能。由图7可以看出,L-J势能均为正值,在液相区形成势垒,势垒高度ΔULJ为液相主体L-J势能与汽相主体L-J势能之差;静电势能均为负值,在液相区形成势阱,势阱深度ΔUe为汽相主体静电势能与液相主体静电势能之差;由于静电势能起主导作用,总势能也为负值,同样在液相区形成势阱,分子之间主要为吸引作用。从图8可以看出,汽相主体势能作用不明显,势垒高度随温度升高而降低,液相主体势能的势阱深度随体系温度的升高而减小。
2.4 模拟分子数对模拟结果的影响
在温度500 K和截断半径rc=0.9498 nm的条件下,当模拟分子数 N=108、256、500和864时,模拟得到的密度分布见图9。统计得到的汽相主体密度ρV、液相主体密度ρL及汽液界面厚度d见表5。

图9 水分子数对密度分布的影响

表5 不同水分子数下界面性质的模拟结果
由表5和图9可见,随着模拟分子数的增加,液相主体密度有所增加,液相主体区域宽度加大,汽液界面厚度稍有增大,汽相主体密度有所波动。
2.5 截断半径对模拟结果的影响
在温度为500 K和模拟分子数为864的条件下,当截断半径 rc=0.7915、0.9498、1.2660 nm时,模拟得到的密度分布如图10所示。统计平均得到的汽相主体密度ρV、液相主体密度ρL及汽液界面厚度d如表6所示。从表6和图10可以看出,随着截断半径的增加,液相主体密度增大,汽相主体密度减小,汽液界面厚度变化不大。

表6 不同截断半径下的水汽液界面性质模拟结果

图10 截断半径对密度分布的影响
3 结论
采用SPC模型,对水汽液界面特性的分子动力学模拟研究结果表明,随着温度的升高,汽相主体密度增加,汽液界面厚度增大,液相主体密度降低,界面张力逐渐减小,液相主体区域势能的势阱深度也逐渐降低。随着模拟分子数的增加,液相主体密度增加,汽液界面厚度稍有增大。随着截断半径的增加,液相主体密度增加,汽液界面厚度变化不大。
[1]李印实,何雅玲,孙 杰,等.不同水分子模型凝结系数的分子动力学模拟对比研究[J].西安交通大学学报,2006,40(11):1272 -1275.
[2]陈正隆,徐为人,汤立达.分子模拟的理论与实践[M].北京:化学工业出版社,2007.
[3]王蕴博.Mg(OH)2粉体干燥动力学及水分子动力学模拟[D].大连:大连理工大学,2012.
[4]Jorgensen W L,Chandrasekhar J,Madura J D,et al.Comparison of simple potential functions for simulating liquid water[J].J Chem Phys,1983,79(2):926 -935.
[5]Mahoney M W,Jorgensen W L.A five-site model for liquid water and the reproduction of the density anomaly by rigid,nonpolarizable potential function[J].J Chem Phys,2000,112(20):8910 -8922.
[6]Rapaport D C.The art of molecular dynamics simulation[M].Cambridge:Cambridge University Press,1995.
猜你喜欢
——《势能》
文化纵横(2022年3期)2022-09-07
天津大学学报(自然科学与工程技术版)(2022年9期)2022-07-04
中学生数理化·八年级物理人教版(2022年6期)2022-06-05
中学生数理化·八年级物理人教版(2021年6期)2021-11-22
环境保护与循环经济(2021年7期)2021-11-02
食品安全导刊(2021年20期)2021-08-30
中学生数理化·八年级物理人教版(2019年6期)2019-06-25
中国科技纵横(2019年5期)2019-04-24
化学反应工程与工艺(2015年1期)2015-04-16
河北工业科技(2015年4期)2015-02-27
