
肌萎缩侧索硬化中病理性TDP-43蛋白研究进展
2012-08-11 08:25:38冯俊强张立志光综述江新梅审校
中风与神经疾病杂志 2012年1期
冯俊强, 张立志, 杨 光综述, 江新梅审校
肌萎缩侧索硬化(ALS)是一种神经系统变性疾病(neurological degenerative diseases)。变性疾病大部分是异质性的,而基因突变造成神经元或胶质细胞内的异常蛋白表达和聚集却常常是共性的。如阿尔茨海默病时的β样淀粉样蛋白变性及微管相关蛋白(tau)病理即为老年斑及神经原纤维缠结主要组成成分即淀粉样原纤维蛋白聚集物[1],路易体(Lewy)中的a-共核蛋白也是构成帕金森病的病理基础。近来神经病理学发现:在额叶变性时出现的泛素包涵体(ubiquitin inclusion,UBI)[2],在 ALS 中也存在[3]。这些异常的蛋白聚集物(包涵体)已经成为了疾病的特异性标志,甚至也是疾病的成因。
2006年,Virginia Lee等发现:在肌萎缩侧索硬化(ALS)及额颞叶痴呆(FTLD)患者的病理性包涵体中检测到TDP-43(trans active response DNA bind ing protein gene分子量为43千道尔顿的具有免疫活性/反应性的DNA结合蛋白)。病理性的TDP-43是一种高磷酸化泛素,其羧基末端只影响中枢神经系统,包括海马、大脑皮质和脊髓。常见于各种神经系统变性疾病[4]。而TDP-43发现于1995年,该蛋白曾是囊肿性纤维化和HIV领域的研究内容。
1 TDP-43的概念及生理功能
TDP-43是由414个氨基酸构成的核蛋白,是由1号常染色体的TARDBP(trans active response DNA-binding protein gene)基因编码的。因其分子量为43 kDa,故被命名为TDP-43。最初发现其在HIV病毒体内的基因组所复制,是一种与TAR-DNA相结合的起到抑制转录的细胞因子[5]。在所有组织中(包括脑组织)呈高度保守及泛素化表达[6,7]。该蛋白含有两个RNA识别序列,在C末端富含甘氨酸。此蛋白是囊性纤维病跨膜调节因子及载脂蛋白A2调节因子复合物[8],在TDP-43中富含甘氨酸的C末端的区域发挥针对外显子的跳跃及拼接的抑制活性[9,10],而且在 TDP-43中富含甘氨酸的C末端可与多种不同类别的核蛋白即异质性的核蛋白(heterogeneous nuclear ribonucleoprotein,hnRNP)进行结合,并与mRNA生成有关[10]。最近又发现,在小鼠的TDP-43可与其临近的SP-10基因(acrosomal vesicle protein 1)启动子结合,参与并调节精子的生成[11]。另外,在发生ALS患者的残存运动神经元中TDP-43可以在其核小体中起到支架作用[12]。正常情况下,TDP-43在胞浆内合成后转变为成熟而稳定的多肽,成熟的TDP-43将会有大部分转运至核内,与TAR DNA结合,发挥调节转录、选择性剪接、参与mRNA的修饰等作用,并起到稳定mRNA的作用,而留在胞浆中的TDP-43含量甚微。在生理状态下,TDP-43多肽经历翻译后修饰,包括磷酸化和泛素化,在发挥完其生理功能后便通过蛋白酶体通路被降解。
2 ALS中的TDP-43
2.1 TDP-43与肌萎缩侧索硬化(ALS)Virginia Lee等研究证明:在对泛素阳性 FTLD[13~17]和 ALS[15,18,19]等疾病时包括神经元胞浆、变性轴突、神经细胞核内包涵体等的研究发现,其中均含有异常的TDP-43,这些TDP-43蛋白在胞浆中异常聚集(正常情况下胞浆中含量甚微),形成类似球形的包涵体,对其进行免疫组化检测,发现其是一种特异且敏感的检测指标。需要指出,在健康的神经细胞和神经胶质细胞中的生理性TDP-43也可以被检测出,而在病理状态下,正常神经细胞核内的TDP-43的表达量显著减少(生理状态下仅在核内发挥作用),而转为胞浆中异常聚集。由于原有的效应含量明显下降,所以在ALS时TDP-43的正常生理功能极有可能是被大大的减损了。需要指出,这些病理性的包涵体同样也呈现泛素化阳性,即在ALS及诸多亚型中的病理性包涵体可同时表现为泛素化阳性及TPD-43阳性。另外,在ALS及其诸多亚型病变中早期无法进行鉴定时,也可以通过对其胶质细胞中的包涵体进行TDP-43的免疫组化技术进行鉴定,以帮助早期诊断病变[15,19,20]。在伴或不伴有痴呆的ALS患者的受累脑组织神经元胞浆及神经胶质细胞内检测到的TDP-43包涵体多呈类球形。大部分ALS病例为散发型病例,仅有大约10%左右为家族型。家族型的ALS在发病的病因上是呈现异质性的,其中约20%是SOD1突变造成的,为常染色体显性遗传[21]。由于在非超氧化物歧化酶基因突变的家族性ALS的患者中,其病理性TDP-43蛋白是明确存在的;而在以SOD1基因突变为病因的一些患者中,其UBI(泛素阳性包涵体)中没有检测到 TDP-43[18,19],这与SOD1基因突变的转基因小鼠模型中没有检测到TDP-43这一结论是相符合的[22],所以有人认为TARDBP突变导致的病理性的TDP-43是非超氧化物歧化酶(SOD1)基因突变的家族型ALS的候补基因。另外,在西太平洋岛屿中的查莫罗人所患的关岛型ALS及以大量tau蛋白为病理表现的帕金森-痴呆症的患者脊髓中检测到了TDP-43[23],在这些患者中的大脑皮质及边缘系统中也检测到了TDP-43[23,24]。这也进一步提示了病理性TDP-43途径是完全不同于SOD1基因突变的一类新的发病途径,当然,这一结论也尚存争议[25]。
2.2 ALS中的TDP-43病理表现 Piao等[26]对102例ALS剖检:发现全部病例的脊髓前前角细胞胞浆中均有包涵体。针对该包涵体进行TDP-43免疫组化染色发现其全部呈现阳性。日本弘前大学神经病理学者[27]对20例ALS患者(其中6例为伴痴呆的ALS,即ALS-D)的皮质、皮质下、新纹状体、边缘系统、颞叶、脊髓等神经组织进行了TDP-43的免疫组化研究,其结果发现:在患者的运动神经元、额颞叶浅部皮质、新纹状体、苍白球、黑质、丘脑等处均有泛素阳性包涵体,其TDP-43也呈阳性反应。在针对皮质、新纹状体、黑质等的神经组织进行的HE染色、单/多克隆TDP-43抗体双重免疫组化染色发现:(1)新纹状体变性程度较皮质、黑质的神经变性轻微;(2)颞叶、黑质的神经元丢失多见于伴有痴呆的ALS(ALS-D),新纹状体神经元丢失并不显著;(3)所有神经组织包括神经胶质细胞均呈TDP-43阳性反应,其中尤以多克隆TDP-43抗体的阳性反应比例高,反应阳性的包涵体位于胞浆中。(4)在ALS-D患者中其TDP-43阳性率高于普通型ALS。(5)包涵体形态分为:大神经元中的枝芽状(见图1)及多形状两种包涵体(见图2),小神经元中的新月形(见图3)或环状(见图4)两种包涵体。而其中大神经元中的枝芽状包涵体仅呈泛素阳性反应,TDP-43呈阴性。以上包涵体围绕细胞核分布或在胞核附近分布。(6)神经胶质细胞内的TDP-43包涵体仍然呈类似表现(见图5~图7)。
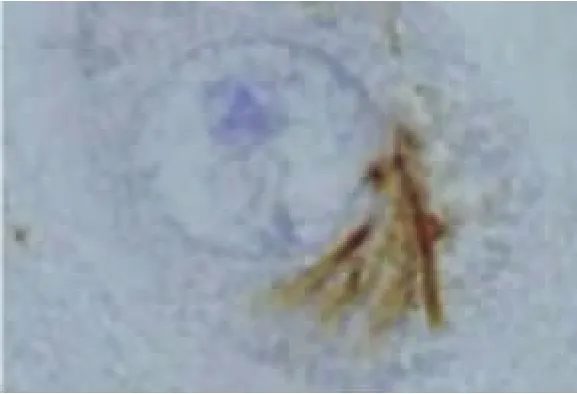
图1 新纹状体大神经元泛素阳性包涵体(标尺5μm)
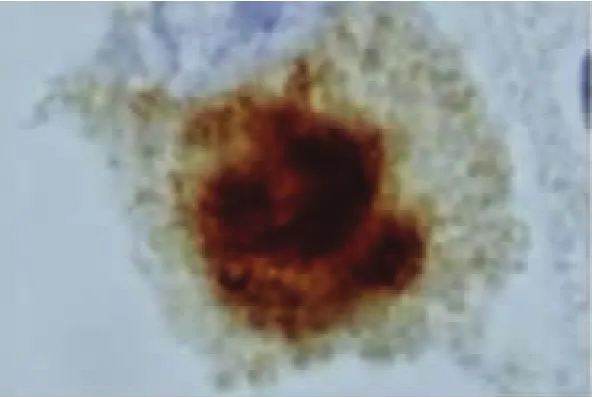
图2 新纹状体大神经元TDP-43包涵体(标尺5μm)

图3 新纹状体小神经元TDP-43包涵体(标尺5μm)
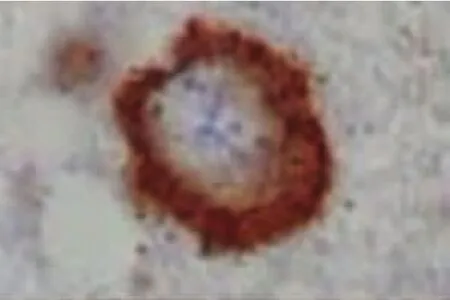
图4 新纹状体小神经元TDP-43包涵体(标尺5μm)
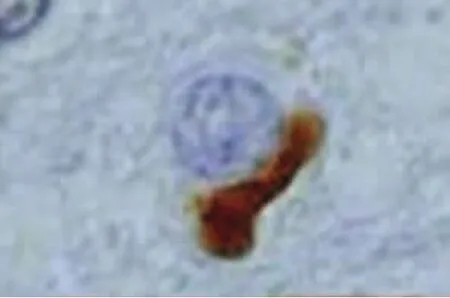
图5 新纹状体神经胶质细胞TDP-43包涵体(标尺5μm)
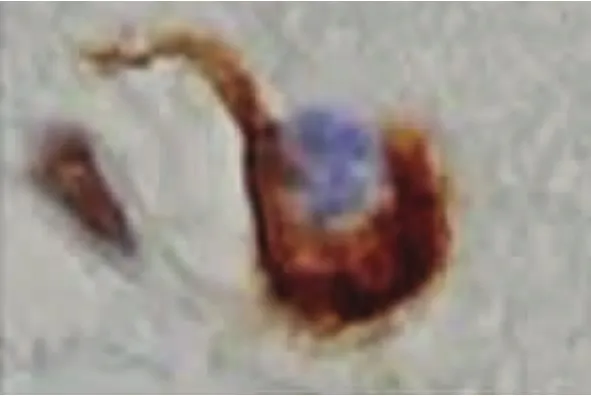
图6 新纹状体神经胶质细胞TDP-43包涵体(标尺5μm)
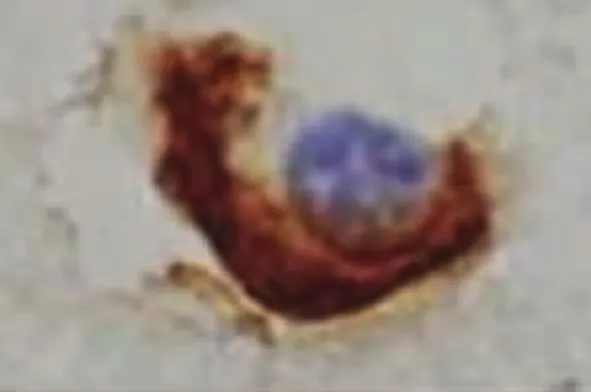
图7 新纹状体神经胶质细胞TDP-43包涵体(标尺5μm)
2.3 病理性TDP-43的分子生物学 由于在伴痴呆的ALS患者中发现了病理性的TDP-43,并且存在于ALS和FTLD-U时的泛素阳性包涵体(UBI)中,TDP-43是二者共有的、相互交织的,在造成神经变性方面起到了同一角色作用。时至今日,TARDBP基因变异已经引人关注[28]。尽管已经发现TDP-43的功能是抑制转录及剪接控制因子[5~7],但是,这种蛋白装置是如何引起神经系统变性的,目前还尚不清楚。至2008年,被鉴定出的TARDBP基因的突变体有多达15种,该基因编码TDP-43,与FTLD-U、家族性和散发ALS患者中神经组织异常蛋白增长有关[29]。诸如:密码子290的甘氨酸(Gly)被丙氨酸(Ala)取代和密码子298的甘氨酸(Gly)被丝氨酸(Ser)取代,而这一区域是TDP-43中富含甘氨酸的区域,此区恰恰具有调控基因表达和调节核蛋白之间的作用功能。又如TARDBP基因外显子6的突变,密码子315的丙氨酸(Ala)被苏氨酸(Thr)替代、密码子337的甲硫氨酸(Met)被缬氨酸(Val)替代,这种病变在两个家系中被鉴定出来,另外,在一些散发ALS患者中的331号密码子谷氨酰胺(Gln)被赖氨酸(Lys)替代、密码子294甘氨酸(Gly)被丙氨酸(Ala)替代[30]。基于此,就形成了一些假说。由于氨基酸被异常的替换,原有结构的稳定性遭到破坏或功能发生错误改变等,使得在包涵体中的TDP-43多价螯合,发生功能缺失,所以造成了转录起始阶段的mRNA出现了异常转录和拼接紊乱。正常情况下TDP-43是一种稳定的低分子量的神经微丝mRNA结合蛋白,与3’UTR(不翻译区)相互作用[31],而在ALS患者中,发现其活性丢失,成为了不稳定的低分子量神经微丝mRNA结合蛋白,其亚基上的化学性质发生改变,造成了神经微丝的聚集而发病。另外,TDP-43的多价螯合,也会导致运动神经元存活基因(SMN)及核内不均一核糖核蛋白(hnRNP)在细胞内的正常分布发生改变,但是这种表达的改变及hnRNP的翻译后修饰的改变在FTLD-U和ALS中并没有被观察到,并且在UBI中也没有检测到hnRNP[32]。此外,包涵体中TDP-43与剪接控制功能有关的C末端发生聚集后,使其正常的生物学活性发生紊乱,也会引起发病,此过程被认为毒性获得功能。另外也有人认为PGRN基因编码TDP-43的配体蛋白,与 TDP-43的细胞核转运有关[33]。在PGRN的功能失调或异常就可导致TDP-43的异常分布。最后,由于TDP-43的异常磷酸化,将导致其信号转导功能的断裂或直接造成TDP-43自身的运输障碍,因此引发神经功能障碍。目前,由于鼠模型的建立及细胞培养技术的进步,正逐步验证上诉假说并阐明TDP-43在ALS及FTLD-U的发病机制中所起的作用。
[1]Forman MS,Trojanowski JQ,Lee VM.Neurodegenerative diseases:a decade of discoveries paves the way for therapeutic breakthroughs[J].Nat Med,2004,10(10):1055 -1063.
[2]Kumar-Singh S,Van Broeckhoven C.Frontotemporal lobar degeneration:current concepts in the light of recent advances[J].Brain Pathol,2007,17(1):104 - 114.
[3]Xiao S,McLean J,Robertson J.Neuronal intermediate filaments and ALS:a new look at an old question[J].Biochim Biophys Acta,2006,1762(11-12):1001-1012.
[4]Neumann M,Sampathu DM,Kwong LK,et al.Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis[J].Science,2006,314(5796):130 -133.
[5]Forman MS,Trojanowski JQ,Lee VM.TDP-43:a novel neurodegenerative proteinopathy[J].Curr Opin Neurobiol,2007,17(5):548 -555.
[6]Buratti E,Baralle FE.Characterization and functional implications of the RNA binding properties of nuclear factor TDP-43,a novel splicing regulator of CFTR exon 9[J].Biol Chem,2001,276(39):36337 -36343.
[7]Ayala YM,Pantano S,D’Ambrogio A,et al.Human,Drosophila and C.elegans TDP-43:nucleic acid binding properties and splicing regulatory fun ction[J].Mol Biol,2005,348(3):575 - 588.
[8]Mercado PA,Ayala YM,Romano M,et al.Depletion of TDP-43 overrides the need for exonic and intronic splicing enhancers in the human apoA-II gene[J].Nucleic Acids Res,2005,33(18):6000 -6010.
[9]Wang HY,Wang IF,Bose J,et al.Structural diversity and functional implications of the eukaryotic TDP gene family[J].Genomics,2004,83(1):130-139.
[10]Buratti E,Brindisi A,Giombi M,et al.TDP-43 binds heterogeneous nuclear ribonucleoprotein A/B through its C-terminal tail:an important region for the inhibition of cystic fibrosis transmembrane conductance regulator exon 9 splicing[J].Biol Chem,2005,280(45):37572-37584.
[11]Acharya KK,Govind CK,Shore AN,et al.cis-Requirement for the maintenance of round spermatid-specific transcription[J].Dev Biol,2006,295(2):781 - 790.
[12]Wang IF,Reddy NM,Shen CK.Higher order arrangement of the eukaryotic nuclear bodies[J].Proc Natl Acad Sci USA,2002,99(21):13583-13588.
[13]Cairns NJ,Neumann M,Bigio EH,et al.TDP-43 in familial and sporadic frontotemporal lobar degeneration with ubiquitin inclusions[J].Am J Pathol,2007,171(1):227 -240.
[14]Davidson Y,Kelley T,Mackenzie IR,et al.Ubiquitinated pathological lesions in frontotemporal lobar degeneration contain the TAR DNA-binding protein,TDP-43[J].Acta Neuropathol,2007,113(5):521-533.
[15]Arai T,Hasegawa M,Akiyama H,et al.TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis[J].Biochem Biophys Res Commun,2006,351(3):602 -611.
[16]Seelaar H,Schelhaas HJ,Azmani A,et al.TDP-43 pathology in familial frontotemporal dementia and motor neuron disease without Progranulin mutations[J].Brain,2007,130(5):1375 -1385.
[17]Higashi S,Iseki E,Yamamoto R,et al.Appearance pattern of TDP-43 in Japanese frontotemporal lobar degeneration with ubiquitin-positive inclusions[J].Neurosci Lett,2007,419(3):213 - 218.
[18]Tan CF,Eguchi H,Tagawa A,et al.TDP-43 immunoreactivity in neuronal inclusions in familial amyotrophic lateral sclerosis with or without SOD1 gene mutation[J].Acta Neuropathol,2007,113(5):535-542.
[19]Mackenzie IR,Bigio EH,Ince PG,et al.Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations[J].Ann Neurol,2007,61:427 -434.
[20]Neumann M,Mackenzie IR,Cairns NJ,et al.TDP-43 in the ubiquitin pathology of frontotemporal dementia with VCP gene mutations[J].J Neuropathol Exp Neurol,2007,66(2):152 -157.
[21]Gros-Louis F,Gaspar C,Rouleau GA.Genetics of familial and sporadic amyotrophic lateral sclerosis[J].Biochim Biophys Acta,2006,1762(11-12):956 -972.
[22]Robertson J,Sanelli T,Xiao S,et al.Lack of TDP-43 abnormalities in mutant SOD1 transgenic mice shows disparity with ALS[J].Neurosci Lett,2007,420(2):128 -132.
[23]Geser F,Winton MJ,Kwong LK,et al.Pathological TDP-43 in parkinsonism-dementia complex and amyotrophic lateral sclerosis of Guam[J].Acta Neuropathol,2008,115(1):133 -145.
[24]Hasegawa M,Arai T,Akiyama H,et al.TDP-43 is deposited in the Guam parkinsonism-dementia complex brains[J].Brain,2007,130(5):1386-1394.
[25]Rothstein JD.TDP-43 in amyotrophic lateral sclerosis:pathophysiology or patho-babel[J]?Ann Neurol,2007,61(5):382-384.
[26]Piao YS,Wakabayashi K,Kakita A,et al.Neuropathology with clinical correlations of sporadic amyotrophic lateral sclerosis 102 autopsy cases examined between 1962 and 2000[J].Brain Pathol,2003,13(1):10-22.
[27]Zhang HX,Tan CF,Mori F,et al.TDP-43-immunoreactive neuronal and glial inclusions in the neostriatum in amyotrophic lateral sclerosis with and without dementia[J].Acta Neuropathol,2008,115(1):115-122.
[28]Rollinson S,Snowden JS,Neary D,et al.TDP-43 gene analysis in frontotemporal lobar degeneration[J].Neurosci Lett,2007,419(1):1-4.
[29]Sreedharan J,Blair IP,Tripathi VB,et al.TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis[J].Science,2008,319(5870):1668-1672.
[30]Van Deerlin VM,Leverenz JB,Bekris LM,et al.TARDBP mutations in amyotrophic lateral sclerosis with TDP-43 neuropathology:a genetic and histopathological analysis[J].Lancet Neurol,2008,7(5):409-416.
[31]Strong MJ,Volkening K,Hammond R,et al.TDP43 is a human low molecular weight neurofilament(hNFL)mRNA-binding protein[J].Mol Cell Neurosci,2007,35(2):320 -327.
[32]Neumann M,Igaz LM,Kwong LK,et al.Absence of heterogeneous nuclear ribonucleoproteins and survival motor neuron protein in TDP-43 positive inclusions in frontotemporal lobar degeneration[J].Acta Neuropathol(Berl),2007,113(5):543 -548.
[33]Ahmed Z,Mackenzie IR,Hutton ML,et al.Progranulin in frontotemporal lobar degeneration and neuroinflammation[J].J Neuroinflammation,2007,4:7.
猜你喜欢
中国运动医学杂志(2016年3期)2016-07-10 12:07:23
中华老年多器官疾病杂志(2016年8期)2016-05-14 07:16:56
中国男科学杂志(2016年9期)2016-03-20 15:00:13
中国组织化学与细胞化学杂志(2016年4期)2016-02-27 11:15:55
磁共振成像(2015年7期)2015-12-23 08:53:04
浙江大学学报(农业与生命科学版)(2015年4期)2015-12-15 12:47:42
结核与肺部疾病杂志(2015年1期)2015-07-18 11:09:22
中国医学科学院学报(2015年5期)2015-03-01 04:03:46
环球中医药(2015年4期)2015-02-27 15:01:51
现代检验医学杂志(2015年2期)2015-02-06 02:01:01
