
阿帕替尼对大鼠肝微粒体细胞色素P4503A1酶活性的影响研究Δ
2012-08-07 03:07梁蔚婷黄红兵冯尧周文菁陈倩超中山大学附属肿瘤医院华南肿瘤学国家重点实验室广州510060
中国药房 2012年9期
梁蔚婷,刘 韬,黄红兵,廖 海,冯尧,潘 莹,魏 雪,周 望,周文菁,陈倩超(中山大学附属肿瘤医院,华南肿瘤学国家重点实验室,广州 510060)
阿帕替尼(Apatinib)是最新研发并已进入Ⅲ期临床的一类新药,是选择性的小分子血管内皮生长因子(VEGF)受体(VEGFR)酪氨酸酶抑制剂,化学名称为甲磺酸N-[4-(氰基环戊基)苯基]{2-[(4-吡啶甲基)氨基](3-吡啶)}甲酰胺(C25H27N5O3S)。药效学研究[1]表明,该药可通过抑制VEGFR酪氨酸酶活性、阻滞VEGF与受体结合后的信号传导、抑制肿瘤血管生成从而治疗肿瘤,在体内外试验中均显示强大的作用。Ⅰ期临床研究显示,阿帕替尼单用或与化疗药物合用都可有效地抑制外源性移植肿瘤物的生成,大部分的抗肿瘤药物都是通过细胞色素P450(CYP)3A4代谢的。考虑到阿帕替尼与其他抗肿瘤药物可能发生的相互作用,本实验在前人研究[2,3]的基础上,在体外孵育体系中运用高效液相色谱-紫外检测(HPLC-UV)法以睾酮作为大鼠CYP3A1的探针底物,定量检测CYP3A1的特异性代谢底物6β-羟基睾酮(6β-OHT)的生成速率,观察阿帕替尼对大鼠肝微粒体CYP3A1酶活性的影响,为研究该药临床应用中与其他药物的相互作用提供依据。
1 仪器与材料
1.1 仪器
Agilent 1100型HPLC系统(美国Agilent公司);Avanti J-30I型高速离心机(德国Beckman公司);XW-80A旋涡混合器(上海医科大学仪器厂);CQ-200超声波清洗器(上海洁净超声设备厂);Ultra-turrax T8组匀浆机(德国IKA公司)。
1.2 试药
阿帕替尼片(江苏恒瑞医药股份有限公司,批号:09051256,规格:每片0.375 g);睾酮(加拿大TRC公司,批号:1-YFD-167-1,纯度:≥98%,分子量:288.42);6β-OHT(批号:051M1145V,纯度:≥97%,分子量:304.42)、氢化可的松(内标,批号:059K1248,纯度:≥98%)、烟酰胺腺嘌呤二核苷酸磷酸酯(NADPH)均购自美国Sigma公司;三羟甲基氨基甲烷(Tris)、乙二胺四醋酸(EDTA)均购自瑞士Roche公司;0.9%氯化钠注射液(浙江济民制药股份有限公司,批号:11071461,规格:每袋250 mL);地塞米松磷酸钠原料药(天津天药药业股份有限公司,批号:DNa090302,纯度:≥97%);BCA蛋白浓度测定试剂盒(上海碧云天生物技术有限公司,批号:0907231116);甲醇(美国Tedia公司);乙酸乙酯、磷酸氢二钠、氯化钙等其他试剂均为国产分析纯。
1.3 动物
SD大鼠25只,♂,SPF级,体重(241±26)g,中山大学实验动物中心提供,动物生产许可证号:SCXK(粤)2009-0011。
2 方法与结果
2.1 色谱条件
色谱柱:Hypersil BDS C18(150 mm×4.6 mm,5 μm);流动相:甲醇-2 mmol·L-1磷酸氢二钠水溶液(pH 8.25,55∶45,V/V),流速:1.0 mL·min-1;检测波长:254 nm;柱温:25 ℃;进样量:20 μL。
2.2 溶液的制备
精密称取睾酮0.288 g,置于5 mL量瓶中,加甲醇溶解并稀释至刻度,摇匀,得浓度为200 mmol·L-1的睾酮母液,置于4℃、备用。精密称取6β-OHT 1 mg,置于5 mL量瓶中,加甲醇溶解并稀释至刻度,得浓度为200 mg·L-1的6β-OHT标准溶液,置于-20℃贮存、备用。精密称取氢化可的松25 mg,置于5 mL量瓶中,用甲醇-水(1∶1)溶解并稀释到刻度,摇匀,得浓度为5 g·L-1的氢化可的松标准溶液,置于4℃贮存、备用。称量适量Tris、EDTA和二硫苏糖醇(DTT),用含20%甘油的双蒸水溶解制成含0.1 mol·L-1Tris、0.1 mmol·L-1EDTA和0.1 mmol·L-1DTT的溶液,用6 mol·L-1的HCl调pH至7.4,制成0.1 mol·L-1Tris-HCl(pH7.4)缓冲液,用于肝微粒体制备。
2.3 钙沉淀法制备大鼠肝微粒体
取大鼠禁食12 h,自由饮水,称重后,断头处死,剖腹,取出肝脏,用4℃0.9%氯化钠注射液冲洗2~3次,直至洗出液为无色或淡黄色,吸干组织并称重,取5 g左右肝组织,以0.25 mol·L-1蔗糖溶液制成20%(W/V)的组织匀浆,16 000 g离心20 min,取上清液转移至超速离心管,加入88 mmol·L-1CaCl2(0.1 mL·mL-1),于冰浴中振摇5 min,27 000 g离心15 min,弃去上清液,沉淀用含20%甘油的0.1 mol·L-1Tris-HCl(pH7.4)缓冲液混悬均匀,分装,于-80℃保存、备用。
2.4 肝微粒体蛋白浓度的检测
取BCA蛋白浓度测定试剂盒中的蛋白标准品(5 mg·mL-1)10 μL,用10 mmol·L-1磷酸盐缓冲液(PBS,pH7.4)溶解并稀释至0.5 mg·mL-1。将0.5 mg·mL-1的蛋白标准品稀释液按0、1、4、8、12、16、20 μL加到96孔板中,用PBS稀释至20 μL(分别相当于浓度为 0、0.025、0.10、0.20、0.30、0.40、0.50 mg·mL-1的蛋白标准溶液)。另外按BCA试剂A-BCA试剂B(50∶1,V/V)制备BCA工作液,在样品孔中加入制备好的BCA工作液200 μL,于37℃恒温箱中静置1 h,于562 nm波长处测定吸光度。以蛋白浓度(X)与吸光度(Y)绘制标准曲线,得回归方程为Y=0.109+1.12X(r=0.998 9)。结果表明,蛋白浓度在0.05~0.50 mg·mL-1线性范围内与吸光度呈良好线性关系(具体步骤参见BCA蛋白浓度测定试剂盒说明书)。
2.5 体外孵育试验
精密吸取20 mmol·L-1的睾酮溶液5 μL,置于10 mL玻璃离心管中,加入适量制备好的肝微粒体,使药物反应体系中肝微粒体蛋白终浓度为0.25 mg·mL-1,用PBS补足体系至450 μL,于 37 ℃水浴中预孵育 5 min,加入 50 μL NADPH(10 mmol·L-1)启动反应,37 ℃水浴10 min,加入3.5 mL冰冷乙酸乙酯终止反应,最后加入10 μL氢化可的松(50 mg·L-1)。
2.6 样品处理
取“2.5”项下终止反应的样品,涡旋1 min,室温放置10 min,3 500 r·min-1离心10 min,吸取上清液转移至另一离心管中,于真空干燥箱中挥干,用甲醇-水(1∶1)200 μL溶解,涡旋1 min,13 000 r·min-1离心10 min,吸取上清130 μL,进样20 μL。
2.7 灭活肝微粒体的制备
取大鼠正常肝微粒体,在95~100℃煮沸1 min,得大鼠灭活肝微粒体。
2.8 专属性考察
分别制备大鼠空白灭活肝微粒体、灭活肝微粒体+睾酮(20 mmol·L-1,5 μL)+氢化可的松(50 mg·L-1,10 μL)+6β-OHT(20 mg·L-1,50 μL)、灭活及正常肝微粒体+睾酮,按“2.5”项下进行孵育后,照“2.6”项下处理,进样分析。结果表明,6β-OHT、氢化可的松、睾酮与其他杂质组分峰分离良好,保留时间分别约为3.94、5.22、14.26 min,且空白灭活肝微粒体中的内源性物质不干扰测定。色谱见图1。
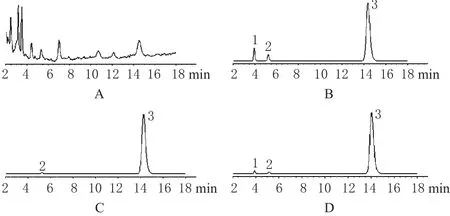
图1 离体高效液相色谱图A.空白灭活肝微粒体;B.灭活肝微粒体+睾酮+氢化可的松+6β-OHT;C.灭活肝微粒体温孵睾酮;D.正常肝微粒体温孵睾酮;1.6β-OHT;2.氢化可的松;3.睾酮Fig 1 HPLC chromatograms in vitroA.blank inactivated liver microsomes;B.inactivated liver microsomes+testosterone+hydrocortisone+6β-OHT standards;C.testosterone incubated with inactivated liver microsomes;D.testosterone incubated with normal liver microsomes;1.6β-OHT;2.hydrocortisone;3.testosterone
2.9 标准曲线的绘制
取10 mL具塞离心管,分别加入5、10、20、40、80、120、160 μg·mL-1的6β-OHT溶液50 μL,加入适量肝微粒体制备成6β-OHT浓度为0.5、1、2、4、8、12、16 μg·mL-1的大鼠灭活肝微粒体温孵样品,按“2.6”项下方法处理,进样测定,以6β-OHT与内标峰面积比(Y)为纵坐标、6β-OHT的浓度(X)为横坐标进行线性回归,得回归方程为Y=0.306 3X+0.064(r=0.999 5)。结果表明,6β-OHT检测浓度的线性范围为0.5~16 μg·mL-1。
2.10 精密度试验
按照“2.9”项下方法制备低、中、高(1、4、12 μg·mL-16β-OHT)浓度的大鼠灭活肝微粒体温孵样品,各5份,按“2.6”项下方法处理,进样测定,每日1次,连续3 d,计算日内和日间RSD,结果见表1。
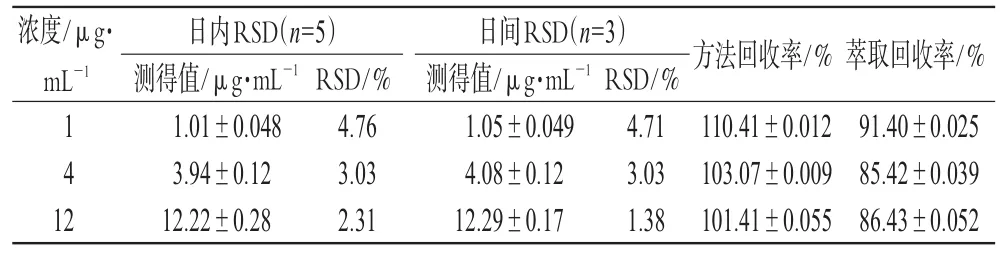
表1 精密度和回收率试验结果Tab1 Results of precision and recovery tests
2.11 回收率试验
取“2.10”项下制备的低、中、高浓度的大鼠灭活肝微粒体温孵样品,各5份,同法操作,以检测值/实际浓度值×100%,计算方法回收率。另取低、中、高浓度的大鼠灭活肝微粒体温孵样品,各5份,处理后进样测定,记录6β-OHT的峰面积A1;再以甲醇-水(1∶1)稀释上述3种浓度的6β-OHT标准溶液,进样测定,其峰面积为A2,6β-OHT在肝微粒体中的萃取回收率以公式A1/A2×100%进行计算。结果见表1。
2.12 稳定性考察
取“2.10”项下制备的低、中、高浓度的大鼠灭活肝微粒体温孵样品,各5份,于室温放置0、4、8 h后,按“2.6”项下方法处理,进样测定。结果,RSD均<5.42%,表明样品在8 h内稳定。
2.13 CYP3A1酶活性研究
2.1 3.1 分组与给药。取25只大鼠随机分为空白对照组(生理盐水1 mL·kg-1)、地塞米松组(100 mg·kg-1)和阿帕替尼高、中、低剂量组(100、50、25 mg·kg-1),每组5只,灌胃给予相应药物,每日1次。除地塞米松组连续给药3 d外,其余各组连续给药14 d。所有大鼠在整个实验期间均自由饮食。
2.1 3.2 给药剂量的选择。阿帕替尼临床应用的推荐剂量是750 mg·d-1[4],按照人体体表面积及物种给药剂量换算公式[5],可计算出大鼠的正常给药剂量为78.125 mg·kg-1·d-1,在预实验中观察到大鼠对150 mg·kg-1·d-1和100 mg·kg-1·d-1的剂量不耐受,因此在设定给药方案时,参考阿帕替尼临床试验(Ⅰ期)报告,大鼠的等效有效剂量为50 mg·kg-1·d-1,以该剂量为中剂量,高低剂量分别递增和递减1倍。阿帕替尼的临床用药周期为1个疗程28 d,因为酶诱导与基因转录增加有关,在4~14 d达到最大值[6],所以本实验将阿帕替尼的给药周期设定为14 d。
2.1 3.3 指标检测。各组大鼠末次给药后禁食不禁水12 h,按“2.3”项下方法取肝微粒体检测其中6β-OHT的浓度。
2.1 3.4 数据处理及结果。关于酶动力学参数的测定,最佳孵育时间及蛋白浓度均需在线性范围内,酶浓度和孵育时间在线性范围内要求底物被结合量低于20%[6],因此,在进行睾酮在大鼠肝微粒体中的酶浓度和孵育时间优化时,发现大鼠肝微粒体孵育体系在2.5~20 min内6β-OHT的产量与时间呈线性关系,结合前述要求,选用了10 min为最佳孵育时间。用探针代谢物6β-OHT的生成速率(6β-OHT的摩尔浓度/孵育时间/肝微粒体蛋白浓度)表示CYP3A1酶活性,各组间的比较采用单因素方差分析,用SPSS 13.0统计学软件处理,结果见表2。
表2 各组6β-OHT的生成速率比较(±s,n=5)Tab 2 Comparison of the productive velocity of 6β-OHT in each group(±s,n=5)
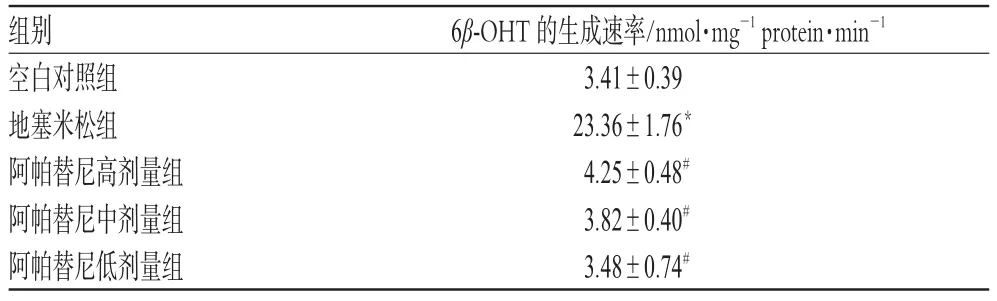
表2 各组6β-OHT的生成速率比较(±s,n=5)Tab 2 Comparison of the productive velocity of 6β-OHT in each group(±s,n=5)
与空白对照组比较:*P<0.01;与地塞米松组比较:#P<0.01vs.blank control group:*P<0.01;vs.dexamethasone group:#P<0.01
6β-OHT的生成速率/nmol·mg-1protein·min-1 3.41±0.39 23.36±1.76*4.25±0.48#3.82±0.40#3.48±0.74#组别空白对照组地塞米松组阿帕替尼高剂量组阿帕替尼中剂量组阿帕替尼低剂量组
由表2可见,阿帕替尼高、中、低剂量组酶活性远低于地塞米松组(P<0.01),与空白对照组无明显差别。因此,本实验结果表明阿帕替尼对大鼠肝微粒体CYP3A1无诱导作用。
3 讨论
CYP450是一组含有亚铁血红素的酶蛋白,又称混合功能氧化酶或单加氧酶,是肝脏中主要的药物代谢Ⅰ相酶,涉及药物代谢的 CYP450酶系主要为 CYP1、CYP2、CYP3 家族,而CYP3A4是其中重要的药物代谢酶,参与50%上市药物的Ⅰ相代谢[7],其在大鼠中的相应亚型为CYP3A1。地塞米松是CYP3A4酶的经典的诱导剂,对人及大鼠的CYP3A4/1均有较强的诱导作用,在考察受试药对CYP3A4酶的诱导作用时,可以地塞米松作为阳性诱导对照,把地塞米松溶于生理盐水对大鼠灌胃给药,剂量为100 mg·kg-1,每日1次,连续3 d[6]。
根据阿帕替尼同类药物的相关研究显示,厄洛替尼、伊马替尼、吉非替尼、索拉非尼均是CYP3A4的底物[8~10],而CYP3A4强抑制剂可以抑制厄洛替尼代谢,使其血药浓度升高[11,12]。伊马替尼的药-时曲线下面积(AUC)可被CYP3A4的酶诱导剂或抑制剂影响而降低或升高[9],索拉非尼和舒尼替尼可抑制CYP3A4酶介导的底物代谢[13],在体外肝微粒体孵育实验中,吉非替尼与厄洛替尼可诱导CYP3A4底物咪达唑仑的代谢,但两者的作用存在差异[14]。由此可推测,阿帕替尼可能会发生由CYP3A4介导的药物相互作用。
多种抗肿瘤药物的代谢,如肿瘤化疗过程中环磷酰胺、紫杉醇、长春新碱等化疗药物都是CYP3A4/5的底物[15,16],阿帕替尼与抗肿瘤药物及其他辅助用药如中药复方制剂合用的现象不可避免,大多数抗肿瘤药物治疗窗狭窄,而CYP3A4参与了临床多种抗肿瘤药物的代谢,CYP3A4酶活性的改变会引起其相应底物的代谢变化,导致相互作用,引起治疗失败或者严重不良反应;同时,中药复方制剂含多种化学成分,与该类药物合用时,发生药物相互作用的可能性就会增加[17,18]。
目前为止,尚无阿帕替尼对肝微粒体CYP450及其主要药物代谢亚型的影响的文献报道,本研究希望探索阿帕替尼在临床应用中,与抗肿瘤药物及其他辅助用药广泛联合应用时,对于重要药物代谢酶CYP3A4活性的影响。本实验结果表明,阿帕替尼对大鼠肝微粒体CYP3A1的活性无诱导作用,但由于大鼠和人的CYP酶存在种属差异,阿帕替尼与抗肿瘤药物及其他辅助用药合用的潜在不良药物相互作用还需要进一步研究。
[1]Tian S,Quan H,Xie C,et al.YN968D1 is a novel and selective inhibitor of vascular endothelial growth factor receptor-2 tyrosine kinase with potent activity in vitro and in vivo[J].Cancer Sci,2011,102(7):1 374.
[2]张 荣,刘昌辉,王宁生,等.以睾酮为探针采用高效液相色谱法测定细胞色素CYP4503A4的酶活性[J].色谱,2008,26(1):80.
[3]黄红兵,刘 韬,邓 多,等.紫杉醇对大鼠肝微粒体CYP3A1的作用[J].中国医院药学杂志,2008,28(24):2 078.
[4]Li J,Zhao XM,Chen L,et al.Safety and pharmacokinetics of novel selective vascular endothelial growth factor receptor-2 inhibitor YN968D1 in patients with advanced malignancies[J].BMC Cancer,2010,10(1):529.
[5]黄继汉,黄晓晖,陈志扬,等.药理试验中动物间和动物与人体间的等效剂量换算[J].中国临床药理学与治疗学,2004,9(9):1 069.
[6]曾 苏.药物代谢学[M].第1版.杭州:浙江大学出版社,2008:221.
[7]Zhou SF.Drugs behave as substrates,inhibitors and inducers of human cytochrome P4503A4[J].Curr Drug Metab,2008,9(4):310.
[8]Li X,Kamenecka TM,Cameron MD.Cytochrome P450-mediated bioactivation of the epidermal growth factor receptor inhibitor erlotinib to a reactive electrophile[J].Drug Metab Dispos,2010,38(7):1 238.
[9]van Schaik RH.CYP450pharmacogenetics for personalizi-ng cancer therapy[J].Drug Resist Updat,2008,11(3):77.
[10]Lathia C,Lettieri J,Cihon F,et al.Lack of effect of ketoconazole-mediated CYP3A inhibition on sorafenib clinical pharmacokinetics[J].Cancer Chemother Pharmacol,2006,57(5):685.
[11]Rakhit A,Pantze MP,Fettner S,et al.The effects of CYP3A4 inhibition on erlotinib pharmacokinetics:computer-based simulation(SimCYP)predicts in vivo metabolic inhibition[J].Eur J Clin Pharmacol,2008,64(1):31.
[12]Smith NF,Baker SD,Gonzalez FJ,et al.Modulation of erlotinib pharmacokinetics in mice by a novel cytochrome P4503A4 inhibitor,BAS 100[J].Br J Cancer,2008,98(10):1 630.
[13]Sugiyama M,Fujita K,Murayama N,et al.Sorafenib and sunitinib,two anticancer drugs,inhibit CYP3A4-mediated and activate CY3A5-mediated midazolam 1’-hydroxylation[J].Drug Metab Dispos,2011,39(5):757.
[14]Li J,Zhao M,He P,et al.Differential metabolism of gefitinib and erlotinib by human cytochrome P450enzymes[J].Clin Cancer Res,2007,13(12):3 731.
[15]Scripture CD,Figg WD.Drug interactions in cancer therapy[J].Nat Rev Cancer,2006,6(9):546.
[16]Kajita J,Kuwabara T,Kobayashi H,et al.CYP3A4 is mainly responsibile for the metabolism of a new vinca alkaloid,vinorelbine,in human liver microsomes[J].Drug Metab Dispos,2000,28(9):1 121.
[17]Harkey MR,Henderson GL,Gershwin ME,et al.Variability in commercial ginseng products:an analysis of 25 preparations[J].Am J Clin Nutr,2001,73(6):1 101.
[18]von Moltke LL,Weemhoff JL,Bedir E,et al.Inhibition of human cytochromes P450by components ofGinkgo biliba[J].Journal of Pharmacy and Pharmacology,2004,56(8):1 039.
猜你喜欢
中国药学药品知识仓库(2022年10期)2022-05-29
河北医药(2020年2期)2020-03-13
中成药(2019年12期)2020-01-04
天然产物研究与开发(2018年9期)2018-10-08
中外医疗(2017年35期)2018-03-07
中成药(2017年9期)2017-12-19
中国实用医药(2017年28期)2017-10-19
中国当代医药(2017年23期)2017-09-20
中成药(2017年5期)2017-06-13
中国实用医药(2017年12期)2017-06-06
