
碳酸钙原位沉积对单根竹纤维拉伸性能的影响1)
2012-07-02 00:03程海涛陈复明
东北林业大学学报 2012年8期
高 洁 王 戈 程海涛 史 强 陈 红 陈复明
(国际竹藤中心,北京,100102)(美国密西西比州州立大学,密西西比州,MS39762-9601)(国际竹藤中心)
慈竹在我国西南各地分布广泛,资源丰富,纤维纵横比大,壁厚腔小,强度高,是一种优良的纸浆原料,随着竹纤维制品的开发,也可应用于纤维板、纤维增强复合材料、纺织等领域[1-3]。碳酸钙(Ca-CO3)常作纤维填料,用于降低成本,改善纸张及天然纤维增强塑料复合材料等的力学性能[4-5]。目前,填充方式主要有共混法、碳化原位加填法及离子溶液原位反应加填法等;其中,离子溶液原位反应加填法具有设备简单、操作方便、CaCO3上载率高等优点[6]。相关研究表明:温度是CaCO3加填纤维工艺的重要因素,影响碳酸钙晶粒尺寸范围及吸附量[7]。笔者采用离子溶液原位沉淀法,不同温度下制备CaCO3改性慈竹纤维,并通过单根纤维拉伸技术对竹单根纤维的力学性质进行了表征,探讨了CaCO3沉积情况对竹单根纤维力学性能的影响,可为纸张等竹纤维制品的CaCO3填充工艺提供新的评价手段和理论支持。
1 材料与方法
1.1 材料
慈竹纤维:四川长宁县3年生慈竹(Neosinoealamus affinis),取其根部往上1.5~3.0 m 处竹肉部分,经V(H2O2)∶V(HAc)=1∶1混合离析液60℃下浸泡24 h左右,离析出单根纤维,再用自来水漂洗至中性,气干保存。用于拉伸的单根竹纤维长2~4 mm,直径8 ~22 μm。
化学药品:氯化钙(CaCl2)、无水碳酸钠(Na2CO3),乙二胺四乙酸二钠(EDTA-2Na),均购自北京国药集团化学试剂有限公司,分析纯,前2种药品用纯水配成0.1 mol/L的离子溶液;914双组分冷固化环氧胶,购自天津燕海化学有限公司。
主要设备:磁力搅拌器(IKA RCT basic),场发射环境扫描电镜(JEOL JSM-6301F,FEI XL30-FEGSEM),马弗炉(CARBOLITE CMF 1300),力学性能测试仪(INSTRON 5848,搭配自主研发的高精度短纤维力学性能测试台(SF-I)),激光共聚焦显微镜(Zeiss LSM 510 Meta)。
1.2 方法
1.2.1 改性竹纤维制备
分别在5、15、25、45、65 ℃下,将6 g 慈竹纤维与300 mL浓度为0.1 mol/L的CaCl2溶液以一定速度混合,20 min后添加等量的Na2CO3溶液和一定量分散剂EDTA-2Na,继续搅拌片刻,获取CaCO3改性竹纤维,均气干,在(23±2)℃,湿度(50±10)%环境中保存。未改性慈竹纤维为离析后的单根竹纤维。
1.2.2 碳酸钙沉积情况及竹纤维的形貌表征
采用场发射环境扫描电镜,结合X射线散射能谱,获取25℃下改性纤维表面颗粒成分及其分布情况,并观察改性前后不同竹纤维的表面形态。纤维表面喷15 nm厚碳膜,样品室内真空度抽至小于5×10-4Pa,扫描电压为15 kV 或5 kV。
1.2.3 竹纤维的灰分测量
根据GB-T 742—2008《造纸原料、纸浆、纸和纸板灰分的测定》测纤维的灰分质量分数,每个水平测2次,误差不超过0.2%。
1.2.4 单根竹纤维拉伸测试
样品采用胶球夹持方式,具体制备和测试过程参考文献[8],拉伸速度为 0.048 mm/min,每组测30根纤维。
2 结果与分析
2.1 纳米碳酸钙原位沉积的表征
图1是由X射线散射能谱在背散射电子成像的25℃改性竹纤维上点扫描获取的颗粒元素分析谱图,图2为经X射线散射能谱面扫描获取的25℃下改性纤维表面C、O、Ca 3种元素的分布情况。

图1 纤维上颗粒的元素分析图
图1重元素(如Ca)的电子反射率大,背散射电子图像的亮度高,而包含轻元素(如C、O)的部分图像暗[9],纤维个体呈暗色,表面颗粒亮,且分布均匀。由X射线能谱点分析得知,颗粒仅由C、O、Ca 3种元素构成,根据离子溶液反应产物可确定它不可能是CaO,所以纤维上呈球状堆叠的颗粒为CaCO3。由图2可看出,纤维表面颗粒的轮廓与C、O、Ca 3种元素富集区域吻合。另外,从左下方的Ca元素分布图得知,在该放大倍率下环境扫描电镜观察不到的纳米CaCO3粒子数目较多,且在纤维上分布均匀。

图2 纤维上颗粒的C、O、Ca元素分布图
2.2 改性前后竹纤维的形貌特征
改性处理前后竹纤维的表面形貌如图3所示。在图3中,单根纤维经化学离析后,因脱去了绝大部分木素和部分半纤维素后,表面出现不少微孔。另外还有纹孔和气干形成的纵向褶皱等凹陷部位,它们有利于离子溶液的渗透,也为原位离子反应形成的碳酸钙颗粒提供了附着位置[10]。5℃时,CaCO3多为不规则四面体的纳米单晶粒,附着量少,但仍可见部分纹孔及褶皱部位被填充。这可能与温度较低时CaCO3成核速率和生长速率低,以及纳米碳酸钙溶解度高有关[11]。15℃以后,碳酸钙附着量明显增多,纳米级单晶粒因纹孔和褶皱的形状自然团聚成球形或椭球形,填充了纤维表面的孔隙。这是因为,随着温度升高,构晶离子的扩散速度增加,Ca2+、离子碰撞几率增大,晶体生长速度加快,先出现的CaCO3晶核最终颗粒大。
2.3 碳酸钙填充对单根竹纤维拉伸性能的影响
表1给出了改性前后单根竹纤维的拉伸测试结果以及灰分质量分数。图4是单根竹纤维拉伸应力—应变典型曲线。
由表1可知,与未改性纤维相比,改性纤维的拉伸强度和弹性模量均有所增加,在25℃水平下达到最大值,分别为 1.35、36.3 GPa。方差分析结果表明:样品的拉伸强度和弹性模量组内差异不显著,断裂伸长率组内组间均不显著。经多重比较得知,15℃下改性纤维的拉伸强度差异显著,而在25℃时单根纤维的拉伸强度和弹性模量差异极显著。由图4可看出,改性前后单根竹纤维在拉伸过程中均呈线弹性断裂,但改性纤维的曲线斜率均比未经处理纤维的大,说明经碳酸钙原位填充后,纤维整体的刚性增大。
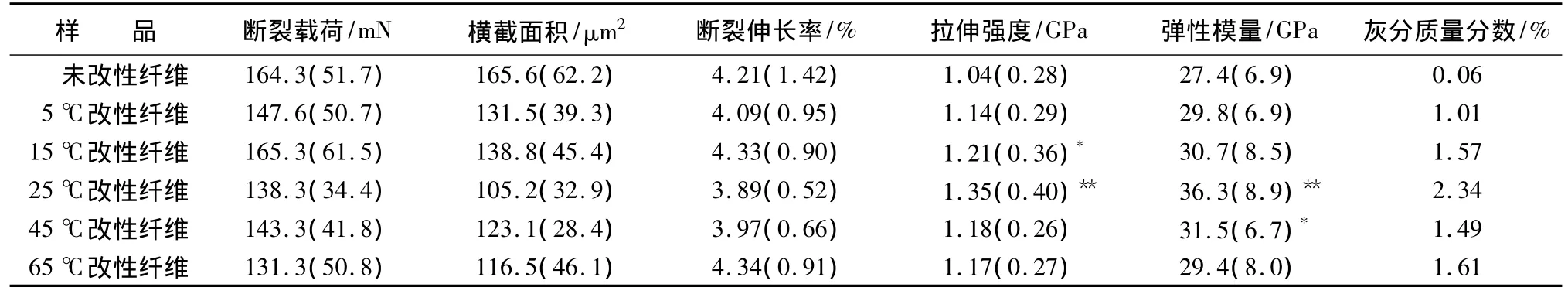
表1 不同单根竹纤维的拉伸和灰分质量分数测试结果

图3 不同竹纤维的表面形貌

图4 不同单根竹纤维的拉伸应力-应变典型曲线
未改性竹纤维的灰分主要是SiO2,含量极少,因此,改性纤维的灰分质量分数间接反映了碳酸钙的上载量。表1表明:随着温度的升高,碳酸钙质量分数先增大再略降低,最终趋近稳定,这与前人的研究结果相一致[7]。另外,纤维的拉伸强度及弹性模量与碳酸钙吸附量的变化趋势相近。同时,碳酸钙的质量分数虽然只占到纤维的1.01% ~2.34%,由图3可知,纳米碳酸钙颗粒原位生长,填充了纹孔、纵向褶皱等纤维本身及制备过程中产生的缺陷部位。Mott等人将环境扫描电镜和拉伸装置联用,动态观察单根纤维的拉伸过程,发现这些缺陷,尤其是在微纤丝角大的纹孔周围,可承受的应力小,最先发生塑性变形;该处又是应力集中部位,成为纤维力学性能的短板,易发生断裂[12]。对于改性纤维,在拉伸过程中,纹孔周围的载荷可传递到填充在内的碳酸钙颗粒上,纹孔构造的迅速形变及破裂可能被有效阻止了,因此,单根纤维的拉伸性能大幅提高。此外,可能由于温度升高后碳酸钙的粒径增大,与纤维的缝隙不再相符合,拉伸过程中承受载荷的作用减小,45℃以后单根竹纤维的拉伸性能有较明显降低,这与冯春等人文献中提到的40℃以后碳酸钙填充纸张的抗拉强度下降趋势一致[7],可以从纸张的承载单元——单根纤维的力学性能变化角度做出解释。
3 结论
随着温度升高,竹纤维上CaCO3附着量呈先增大再减小的趋势,纳米碳酸钙颗粒沿着纤维上纹孔、纵向褶皱等部位的生长速度加快,形貌由单一不规则四面体变为多颗团聚体,呈椭球形或球形,粒径增大。
经CaCO3改性处理后单根慈竹纤维的拉伸强度、弹性模量均提高,25℃改性纤维的性能最佳,分别为1.35、36.3 GPa。这可能由于拉伸过程中填充在竹纤维微孔中的CaCO3颗粒起到了承受载荷,减少应力集中的作用。
将纳米碳酸钙原位沉积到竹纤维上,不仅填充了竹纤维的孔隙,改善了竹纤维的力学强度;同时,纤维上的纳米级、微米级孔径也成为碳酸钙晶粒的生长模板,既为碳酸钙晶核的生长提供了位置,又限制了附着在纤维上的碳酸钙尺寸,起到良好的分散纳米碳酸钙颗粒的效果,若将改性竹纤维用于竹塑复合材料的制备,可能会更好地发挥纳米碳酸钙和竹纤维的增强增韧协同效果,改善竹塑复合材料的力学性能。
[1]何天健.慈竹、黄竹纸浆竹林的培育及造纸研究[J].竹子研究汇刊,1991,10(4):62-71.
[2]王戈,王越平,程海涛,等.我国纺织用竹纤维的研究与开发[J].木材工业,2010,24(4):18-20.
[3]詹永富,蔡建,邱莎莎,等.竹塑复合材料军用包装箱应用研究[J].包装工程,2008,29(12):96-97.
[4]Subramanian R,Fordsmand H,Paulapuro H.Precipitated calcium carbonate-cellulose composite fillers:Effect of PCC particle structure on the production and properties of uncoated fine paper[J].Bioresources,2007,2(1):91-105.
[5]姜锋,秦特夫.木塑复合材料增韧的研究[J].塑料工业,2007,B(6):137-140.
[6]Ciobanu M,Bobu E,Ciolacu F.In-situ cellulose fiber loading with calcium carbonate precipitated by different methods[J].Cellulose Chemistry & Technol,2010,44(9):379-387.
[7]冯春,陈港.制备温度对碳酸钙—细小纤维复合填料性能的影响[J].造纸科学与技术,2010,29(1):33-37.
[8]曹双平,王戈,余雁,等.几种植物单根纤维力学性能对比[J].南京林业大学学报:自然科学版,2010,34(5):87-90.
[9]杜学礼,潘子昂.扫描电子显微镜分析技术[M].北京:化学工业出版社,1986:111-112.
[10]Allan G G,Negri A R,Ritzenthaler P.The microporosity of pulp;the properties of paper made from pulp fibers internally filled with calcium carbonate[J].Tappi Journal,1992,75(3):239-244.
[11]Klungness J H.Fiber loading improvements in papermaking:US,2010/0212853A1[P].2010.
[12]Mott L,Shaler S M,Groom L H.A technique to measure strain distributions in single wood pulp fibers[J].Wood and Fiber Science,1996,28(4):429-437.
猜你喜欢
实用手外科杂志(2022年2期)2022-08-31
选煤技术(2022年2期)2022-06-06
选煤技术(2022年2期)2022-06-06
煤化工(2021年4期)2021-09-13
四川蚕业(2021年4期)2021-03-08
无机盐工业(2021年1期)2021-01-08
石材(2020年12期)2020-12-31
钻井液与完井液(2019年4期)2019-10-10
农业工程学报(2018年15期)2018-08-21
中成药(2018年5期)2018-06-06
