
中国新疆紫花苜蓿复合体3个种的遗传多样性及亲缘关系研究
2012-06-08 08:22李飞飞崔大方羊海军邓超宏李庆艳
草业学报 2012年1期
李飞飞,崔大方,羊海军,邓超宏,李庆艳
(1.华南农业大学生命科学学院,广东 广州510642;2.华南农业大学林学院,广东 广州510642;3.华南农业大学公共基础实验教学中心,广东 广州510642)
苜蓿属(Medicago)植物是豆科(Leguminosae)一个重要的类群,大多数苜蓿属物种都是广泛分布在温带草原上的优质牧草。而紫花苜蓿(M.sativa)、黄花苜蓿(M.falcata)以及多变苜蓿(M.varia)更是畜牧业主要的饲料来源[1-3]。
紫花苜蓿复合体包括9个可以自然杂交的分类群[4],包含有二倍体和四倍体物种。在我国仅分布有3个种,即紫花苜蓿、黄花苜蓿和多变苜蓿。3个种均为苜蓿属多年生草本植物,紫花苜蓿花冠各色,淡黄,深蓝或暗紫色、荚果螺旋2~4圈、中央无孔或近无孔,在全国各地都有栽培或呈半野生状态;黄花苜蓿花冠黄色、荚果镰形,分布于我国东北、华北和西北地区以及中亚及欧洲地区;多变苜蓿是紫花苜蓿与黄花苜蓿自然杂交产生的类型,其花冠浅紫色、黄色或其他、荚果螺旋0.5~2.0圈、中央有孔,在我国新疆和高加索、中亚细亚、西伯利亚一带多有分布[5]。
由于这3个种形态相似,并且长期以来在不同的地理条件下杂交,子代和亲本类型多样,使其分类问题存在很大争议。前苏联学者在20世纪初到50年代把它们分成许多种,而中国、美国、加拿大学者则支持将它们定为一个种即多变苜蓿。在Small和Jomphe[6]系统中将多变苜蓿以及Lesins和Lesins[7]系统中的紫花苜蓿、黄花苜蓿划分为紫花苜蓿种下的3个亚种,分别是M.sativasubsp.varia、M.sativasubsp.sativa及M.sativasubsp.falcata。我国学者何咏松和吴仁润[8]通过形态学特点和遗传现象的观察认为紫花苜蓿和黄花苜蓿属同1个种的2个亚种或生物型或生态型。李拥军和苏加楷[9,10]采用种子贮藏蛋白技术及RAPD(随机扩增DNA多态性,random amplified polymorphic DNA)标记分析发现新疆大叶苜蓿、新牧1号多变苜蓿和新疆黄花苜蓿之间的遗传距离为0,支持将这3个苜蓿类型划分为紫花苜蓿的3个亚种。而王俊杰[11]认为荚果形态在苜蓿属植物“种”及以上分类地位的界定上是十分有效的,而在种下分类单位的界定上则难以发挥作用,因此不支持Small和Jomphe[6]系统中的划分,并且将中国黄花苜蓿野生种群的荚果划分为13个类型。因此,紫花苜蓿、黄花苜蓿及多变苜蓿这3个种及其种下类型的分类地位需进一步研究确定。
近十余年来,许多分子标记包括简单串联重复序列SSR(简单重复序列,simple sequence repeats)[12-14],RAPD[15,16],RFLP(限制性片段长度多态性,restriction fragment length polymorphism)[17]以及细胞器基因片段[18,19]都曾对苜蓿属植物进行过研究。但这些研究中绝大部分以栽培的紫花苜蓿为主要研究材料,对野生分布的多变苜蓿及黄花苜蓿研究较少,而将共显性与显性分子标记结合对广泛分布于新疆地区的紫花苜蓿复合体3个种的研究还未有报道。因此本研究利用SSR及ISSR(简单重复序列区间,inter-simple sequence repeat)分子标记技术,比较分析在中国新疆分布的紫花苜蓿、黄花苜蓿和多变苜蓿3个种共10个居群的遗传多样性及其亲缘关系,为苜蓿属植物的分类学问题以及苜蓿属植物资源的合理开发利用和保护提供分子依据。
1 材料与方法
1.1 实验材料及引物
实验材料于2007-2008年在野外采集,共有3个种10个居群,每个居群选取19份单株,共采集190份单株,记录编号后分别放入装有变色硅胶的封口袋中干燥。居群编号、采集地及采集地经纬度见表1。40对SSR引物来源于Bernadette等[20]发表的紫花苜蓿SSR引物,37个ISSR引物来源于UBC大学公布的100个通用引物。
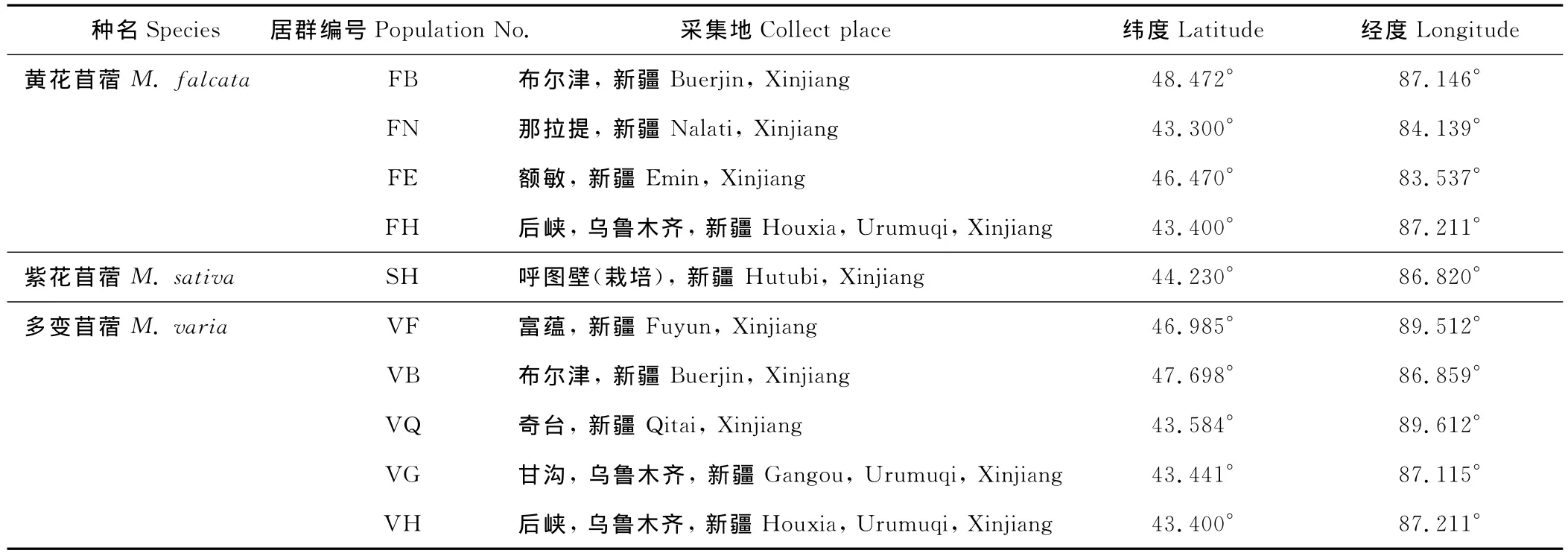
表1 样品采集地信息Table 1 Sample list of location
1.2 实验方法
于2008年将野外采集的苜蓿属植物每份单株摘取适量叶片,采用改良CTAB(hexadecyltrimethy-ammonium bromide)法提取植物总DNA,并用1.0%的琼脂糖电泳检测DNA的浓度和纯度。
SSR反应体系:总体积20μL,包括10×Buffer(含 Mg2-)2μL,10mmol/L dNTP 0.3μL,10μmol/L引物各1μL,Taq酶(5U/μL)0.2μL,30ng/μL DNA 模板2μL,去离子水13.5μL。SSR反应程序:94℃变性5 min;94℃变性30s,54℃退火1min,72℃延伸1min,35个循环;72℃延伸10min;4℃保存。ISSR反应体系:总体积20μL,包括10×Buffer(含 Mg2-)2μL,10mmol/L dNTP 0.25μL,10μmol/L 引物1μL,Taq酶(5 U/μL)0.2μL,30ng/μL DNA模板2μL,去离子水14.55μL。ISSR反应程序:94℃变性5min;94℃变性30s,54℃退火45s,72℃延伸1.5min,42个循环;72℃延伸7min;4℃保存。
SSR扩增产物采用6%的聚丙烯酰胺凝胶电泳检测,ISSR扩增产物采用2%琼脂糖凝胶电泳检测。电泳结果至凝胶成像仪拍照观察,并记录结果。
1.3 数据统计分析
由于紫花苜蓿和多变苜蓿为同源四倍体,而黄花苜蓿既含有四倍体又含有二倍体[21],所以二倍体等位基因的频率统计公式并不适合本研究,刘志鹏等[22]曾采用一种将无效等位基因包含在内的统计方法统计四倍体,但由于这种方法还未完善,部分统计指数还需进一步研究。因此本实验统一采用0/1法统计,以扩增条带在相对迁移位置有无,赋以“1”或“0”,生成分子数据矩阵。利用PopGene 1.32软件[23]进行电泳谱带差异和各项遗传指数的统计分析,包括:多态位点百分率(PPB)、有效等位基因数(ne)、Nei’s基因多样性指数(h)、遗传分化系数(Gst)、基因流(Nm)、Shannon信息指数(I)、遗传距离(GD)。利用 GeneALEx软件[24]AMOVA 计算方差分量,及Mental检测遗传距离和地理距离相关系数(r)。SSR/ISSR引物多态信息含量采用计算公式:

式中,Pi为微卫星位点上第i个等位基因频率(allelic frequency),Pj为第i+1个等位基因频率。利用POPTREE2[26]软件采用非加权平均聚类法(UPGMA)绘制聚类图,并重复10 000次计算自展支持率[27]。
2 结果与分析
2.1 SSR及ISSR位点多态性
40对SSR引物中筛选出15对多态性好、具清晰条带的SSR引物用于进一步研究,在10个居群190份材料中共获得66个位点,64个多态性位点,位点数范围为2(MTIC258)~11(MTIC248),平均位点数达到4.4个,平均多态性位点数达4.3个,多态位点百分比达到96.97%;10个ISSR引物共获得74个位点,多态位点百分比达到100%,位点数范围为2(891)~13(822),平均多态性位点数达7.4个(表2)。
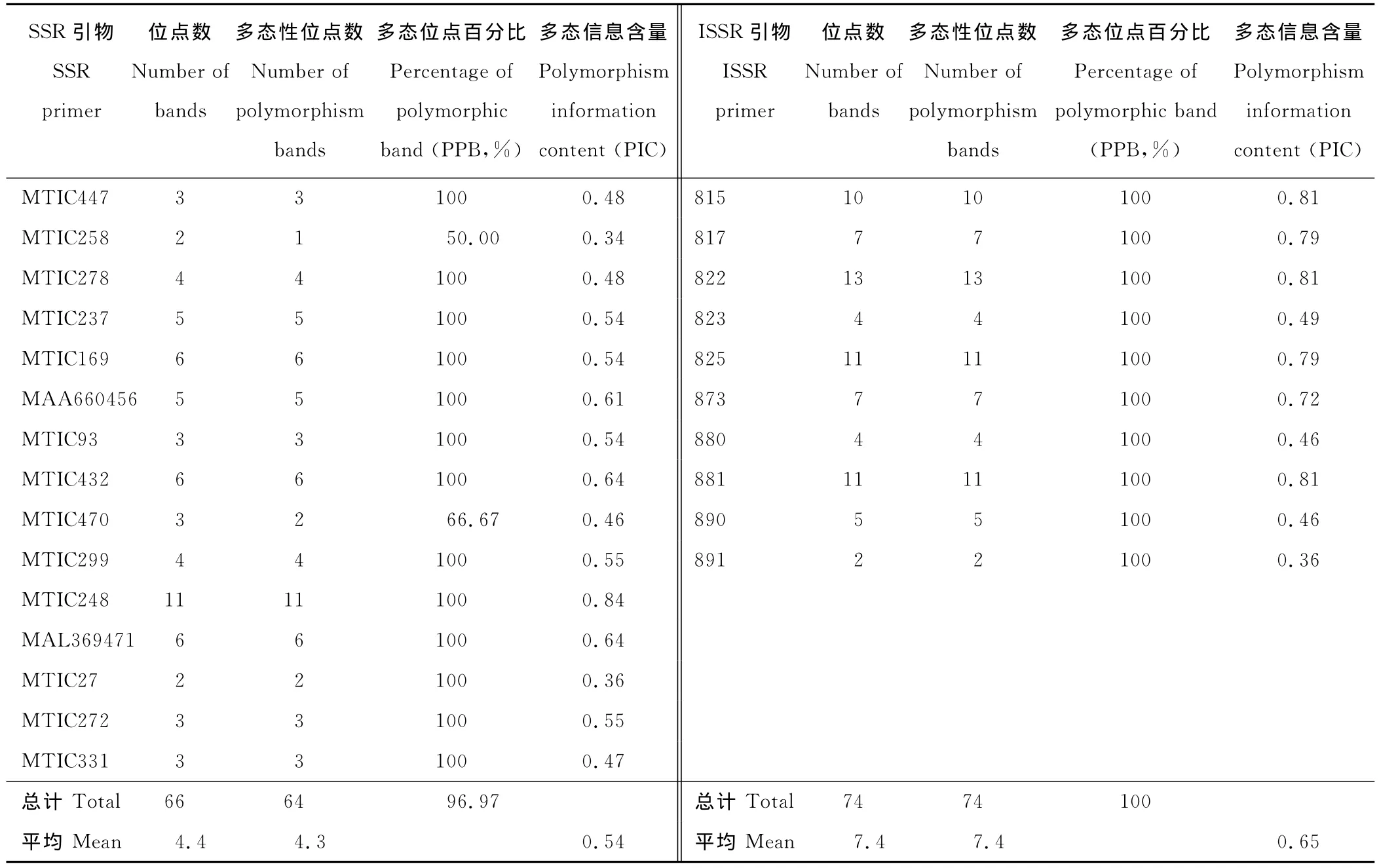
表2 15对SSR引物及10个ISSR引物在总群体中的遗传多样性信息Table 2 Genetic diversity informations of 15pairs of SSR primers and 10ISSR primers in total group
多态信息含量是衡量微卫星位点多态性高低的较好的指标,当PIC>0.50时,具有高度多态性;当0.25<PIC<0.50时,具有中度多态性;当PIC<0.25时,具有低度多态性。SSR标记中15个位点中9个位点具有高度多态性,6个位点具有中度多态性,其中MTIC248引物PIC值最高(0.84),MTIC258引物PIC值最低(0.34),平均值为0.54。ISSR标记中10个位点中6个位点具有高度多态性,4个位点具有中度多态性,其中881引物PIC值最高(0.81),891引物最低(0.36),平均值为0.65。
2.2 紫花苜蓿复合体10个居群遗传多样性分析
SSR标记下10个居群的遗传变异显示(表3),不同居群内的多态位点百分率不同,范围为57.58% (FE)~75.76% (VG),平均值为68.79%,总群体为96.97%;各居群Shannon信息指数为0.24(FE)~0.33(VG),平均值为0.30,总群体为0.35;各居群 Nei’s基因多样性指数为0.16(FE)~0.21(VG),平均值为0.19,10个群体基本接近,变异幅度不大,总群体为0.22。10个居群中多变苜蓿居群VG以上3个指数均最高,可见在SSR分子标记反映的结果中多变苜蓿居群VG遗传多样性最丰富,黄花苜蓿居群FE的3个指数均最低;多变苜蓿5个居群中3个指数最高的为居群VG,最低的为居群VQ,黄花苜蓿4个居群中3个指数最高的为居群FB,最低的为居群FE;多变苜蓿总群体多态位点百分率(92.42%)、Shannon信息指数(0.35)、Nei’s基因多样性指数(0.22)均大于黄花苜蓿总群体指数(PPB=86.36%、I=0.32、h=0.20);紫花苜蓿复合体总群体基因分化系数为0.12,基因流为3.62,多变苜蓿居群间基因流 (7.75)大于黄花苜蓿居群间基因流(5.21)。
ISSR标记下10个居群的多态位点百分率为47.30% (SH)~75.68% (FH),平均值为66.49%,总群体为100%;各居群Shannon信息指数为0.19(SH)~0.30(FH),平均值为0.26,总群体为0.33;各居群 Nei’s基因多样性指数为0.12(SH)~0.19(FH),平均值为0.17,10个群体非常接近,总群体为0.20。与SSR分析结果不同的是10个居群中黄花苜蓿居群FH以上3个指数均最高,紫花苜蓿3个指数均最低;多变苜蓿5个居群中居群VB以上3个指数最高,居群VG最低,黄花苜蓿4个居群中居群FH以上3个指数最高,居群FE最低;多变苜蓿总群体多态位点百分率 (93.24%)大于黄花苜蓿总群体(91.89%),但多变苜蓿总群体Shannon指数(0.30)和 Nei’s基因多样性指数 (0.18)略小于黄花苜蓿(I=0.32,h=0.20);总群体基因分化系数为0.18,基因流为2.34,基因流低于SSR标记的分析结果,但多变苜蓿居群间基因流(5.43)同样高于黄花苜蓿(4.94)。黄花苜蓿居群FH在2种分子标记下都存在特有位点,但在ISSR标记下紫花苜蓿存在1个特有位点。
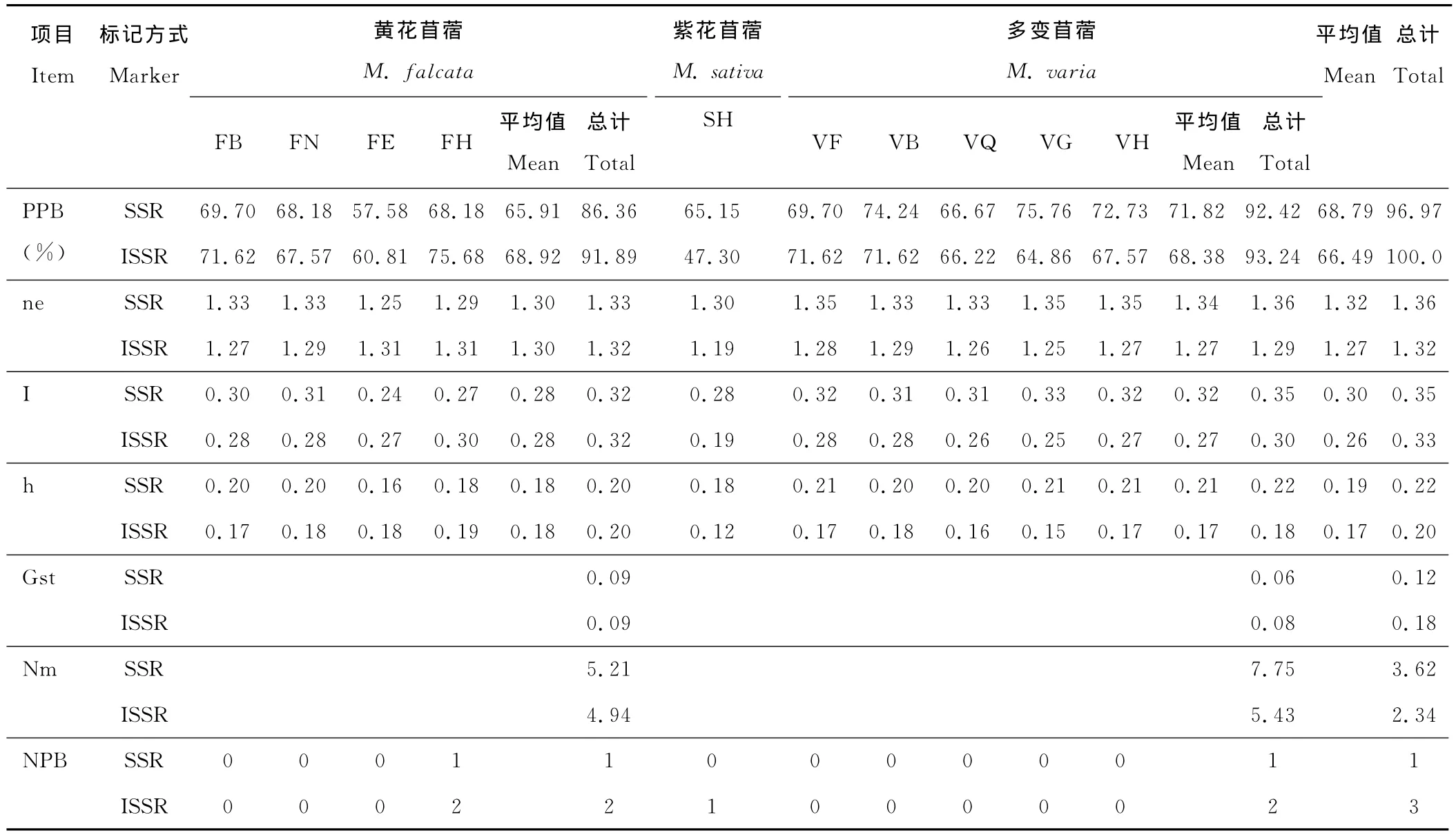
表3 紫花苜蓿复合体10个居群遗传变异比较Table 3 Genetic variation of 10populations of M.sativacomplex
2.3 紫花苜蓿复合体亲缘关系分析
在2种分子标记下总群体Shannon指数 (0.35,0.33)以及Nei’s基因多样性指数 (0.22,0.20)均高于10个居群的平均值(I=0.30,h=0.19;I=0.26、h=0.17),可反映出遗传多样性主要来源于居群内而不是居群间,群体的AMOVA分子方差分析得出SSR标记下遗传变异的7%来源于种间,7%来源于种内居群间,86%来源于居群内,ISSR标记下遗传变异的12%来源于种间,遗传变异的10%来源于种内居群间,79%来源于居群内,2种分子标记均证明遗传变异主要分布在居群内。但2种标记对种间、居群间、居群内差异的划分并不十分明显(表4)。
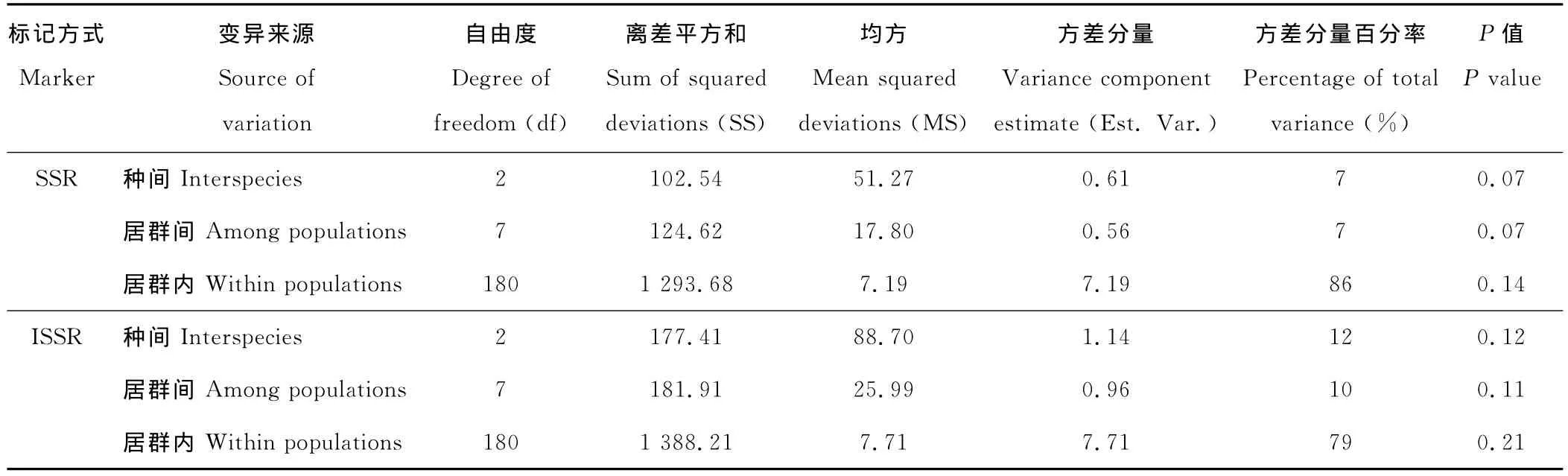
表4 紫花苜蓿复合体10个居群SSR变异的分子方差分析Table 4 Analysis of molecular variance(AMOVA)for 190individual plants using 66SSR alleles and 74ISSR bands respectively
SSR标记中Nei’s遗传距离平均值为0.07,ISSR标记中Nei’s遗传距离平均值为0.19。利用Mantel检测,2种分子标记下10个居群的遗传距离相关系数(r)为0.50,P<0.001,呈显著相关。SSR与ISSR两种分子标记的UPGMA聚类分析表明(图1):3个种中,黄花苜蓿与多变苜蓿关系较近,紫花苜蓿与其他2种遗传距离较远,单独形成一支,自展支持率为100%;紫花苜蓿的所有居群在2种标记下均聚为一类;而黄花苜蓿的FE居群在SSR标记下单独成为一支,且自展支持率为65%。
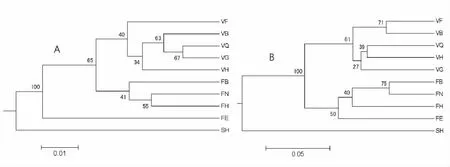
图1 基于SSR(A)及ISSR(B)标记下紫花苜蓿复合体3个种10个居群Nei’s遗传距离构建的UPGMA树状图Fig.1 UPGMA dendrograms of the genetic relationships among 10populations of M.sativacomplex constructed from estimated simple matching genetic distance based on SSR markers(A)and ISSR markers(B)
Sun等[28]认为不同标记体系的数据综合起来进行整体分析,更能客观的反映种间关系。因此,本研究将ISSR和SSR数据合并起来,计算紫花苜蓿复合体10个居群的遗传距离,并构建UPGMA树。结果显示遗传距离范围为0.05(VF和 VB)~0.23(VQ和SH),平均遗传距离为0.12,大于SSR标记下的平均遗传距离,小于ISSR标记下的平均遗传距离。基于2种分子标记构建的UPGMA树(图2)显示,多变苜蓿的所有居群聚为一支,自展支持率为86%。在多变苜蓿的5个居群中进一步划分为2个亚分支,一支包含VQ、VG和VH居群,另一支包含VF和VB居群。黄花苜蓿的3个居群聚为一支,自展支持率为79%,FE居群从其他居群中分离出来。Metal相关性分析证明,多变苜蓿5个居群和黄花苜蓿4个居群的遗传距离与其相应的地理距离(表5)不相关,相关系数(r)分别为0.49,P>0.05;-0.32,P>0.1。
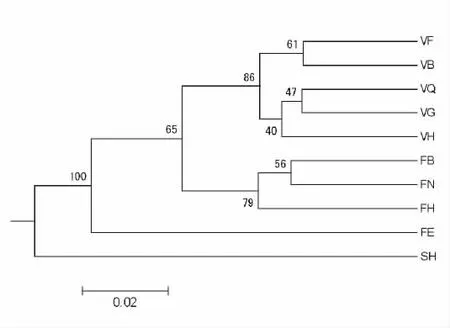
图2 基于全部分子数据的紫花苜蓿复合体3个种10个居群的Nei’s遗传距离UPGMA树状图Fig.2 UPGMA dendrogram of the genetic relationships among 10 populations of 3species constructed from estimated simple matching genetic distance based on SSR and ISSR combined data

表5 黄花苜蓿居群间及多变苜蓿居群间的地理距离Table 5 Geographical distances among populations of M.falcataand M.varia km
3 讨论
3.1 紫花苜蓿复合体3个种及种下居群遗传多样性分析
本研究首次以SSR和ISSR标记对紫花苜蓿复合体居群内及居群间的遗传分化模式进行了探讨,结果发现,2种分子标记的平均多态信息含量都达到了高度多态性,且SSR及ISSR位点在居群内及居群间均表现为丰富的遗传变异,表明居群内及居群间均存在丰富的遗传多样性。此外SSR等位变异与ISSR位点在居群间和居群内的变异程度极不平衡,不同位点间的变异相差较大。结果还发现SSR标记中仅FH具有特有变异位点,ISSR标记中仅居群FH和VH具有特有变异位点,这2个居群的采集地都位于新疆乌鲁木齐后峡距白杨沟10km处,因此推测这个采集地的黄花苜蓿与多变苜蓿具有较丰富的遗传多样性,保存价值大。
在SSR分子标记下的群体遗传多样性分析表明,黄花苜蓿居群FE的多态位点百分率、Shannon信息指数、Nei’s基因多样性指数、PIC指数均最低,并具有最多纯合位点,与其他居群差别较大,分析主要有以下2个原因,1)黄花苜蓿居群FE的样品形态特征为叶片小、黄色花、荚果镰形,且生境偏旱,为黄花苜蓿的变种草原苜蓿(M.falcatavar.romanica)。2)采自额敏喇嘛昭山前荒漠草原,地理分布上处于相对封闭的状态,人口牲畜来往不频繁,不会带来太多的杂交机会。
10个居群相比较的结果表明,SSR标记分析中居群VG遗传多样性最高,FE遗传多样性最低,ISSR标记分析中居群FH遗传多样性最高,紫花苜蓿遗传多样性最低。因此不同的分子标记在分析遗传多样性时所得到的结果有所不同,进一步分析发现VG位于新疆乌鲁木齐甘沟草原站的保护区内,分布区内的植被得到了当地很好的保护,而FH具有特有变异位点,因此这2个居群在不同分子标记中表现出最丰富的遗传多样性具有其合理性。而紫花苜蓿在ISSR标记中表现出较低的遗传多样性,推测与其为栽培品种有关。多变苜蓿与黄花苜蓿总体遗传多样性相差不大,在2种分子标记中高低各有不同,但多变苜蓿多态性比率在2种分子标记中均高于黄花苜蓿,说明多变苜蓿遗传变异略大于黄花苜蓿。
通过种间-种内、居群间-居群内各层次结构的AMOVA分析发现,2种标记均显示群体遗传变异主要来源于居群内。Hilde和Lgor[29]认为,一年生或短命多年生、自交和演替阶段早期的类群在居群间保持有高的遗传变异,而寿命长、异交、演替阶段晚期的类群在居群内保持有高的遗传变异。而紫花苜蓿复合体3个种均为多年生、异交、演替阶段晚期植物,因此在居群内具有高的遗传变异与其本身生物学特性有关。
多变苜蓿5个居群之间的基因流(SSR Nm=7.752,ISSR Nm=5.431)大于黄花苜蓿4个居群之间的基因流(SSR Nm=5.214,ISSR Nm=4.939);SSR标记中总群体基因流为3.619,ISSR标记分析中总群体基因流为2.336。本研究结果中的基因流均大于一般广布种植物的基因流(Nm=1.881)[30],且 Wright[31]认为,当 Nm值大于1时基因流就可以防止由遗传漂变引起的居群之间的分化,因此紫花苜蓿复合体种群中的基因流防止了3个种居群之间的分化,并防止了同源四倍体在进化过程上变得呆板。
虽然基因流对防止物种分化具有很重要的作用,但另一方面,在人类活动干扰或自然环境变化过程中,种间基因流和杂交可能导致原生境隔离的近缘物种间产生杂交没化(hybridization merging),使得物种间的区别不再像以前明显进而导致物种合并[32,33],形成不同分类群间遗传同化(genetic assimilation)现象。因此介于紫花苜蓿复合体3个种之间具有的杂交现象,其种群的基因流会随时间而改变,需定时对其进行测定,以了解当前这3个种的遗传分化。
3.2 紫花苜蓿复合体3个种的亲缘关系分析
从聚类图上分析,SSR与ISSR数据合并构成的聚类图不仅符合2个标记共有的特征,且所反映三者之间的关系更为客观。UPGMA树上形成3个主要的分支,紫花苜蓿独立成一支,来源于同一采集地的黄花苜蓿居群FH与多变苜蓿居群VH都已分开,充分说明多变苜蓿、黄花苜蓿和紫花苜蓿虽然遗传关系较近,但三者之间仍可以得到有效地划分,因此尽管这3个种之间的遗传关系非常近,但本研究仍支持将多变苜蓿、黄花苜蓿和紫花苜蓿划分为3个种。很长一段时间,黄花苜蓿是否应当划分为一个单独的种都存在争议。Brummer等[34]采用RFLP分析二倍体和四倍体苜蓿发现黄花苜蓿与M.caerulea不同,能有效的区分。Havananda等[35]基于叶绿体和线粒体基因同样也讨论了黄花苜蓿与M.caerulea的关系,叶绿体的结果显示黄花苜蓿明显的与较原始的种M.caerulea分别开来,而线粒体的结果却不明显。因四倍体的紫花苜蓿是由M.caerulea直接进化而来,故本研究中基于SSR和ISSR两种分子标记分析的结果与Havananda等[35]的研究结果相似。而孙毅等[36]利用核基因ITS的研究结果显示黄花苜蓿与紫花苜蓿应该合并为一个种。因此,这些研究结果并不能决定这3个分类群为种或亚种,但可以确定的是他们的DNA序列很不相同,利用不同的基因片段分析得出的结果会不一样。从地理分布和物种特性上来看,完全野生的紫花苜蓿没有记载,而黄花苜蓿多为野生分布。因此本研究支持将这3个分类群作为3个种的划分。遗传关系显示多变苜蓿与黄花苜蓿关系较近,这与李拥军和苏加楷[10]利用RAPD分子标记所得到的结果有所不同,他认为多变苜蓿与紫花苜蓿遗传距离更小。
多变苜蓿、黄花苜蓿各居群的遗传距离与其地理距离相关性分析发现没有显著相关性存在于多变苜蓿及黄花苜蓿的遗传距离与地理距离之间。这主要由于紫花苜蓿各居群的生长环境不一致导致,而黄花苜蓿居群FE主要为草原苜蓿与其他居群遗传分化较大,明显区分于其他居群分支而单独存在。因此本研究的分子标记所得到的结果支持Lesins和Lesins[7]的结论,即草原苜蓿为黄花苜蓿一个变种的分类学地位。
通过以上研究,得出以下3个结论:1)多变苜蓿居群VG和黄花苜蓿居群FH在10个居群中遗传多样性最高,并且居群FH及居群SH含有特有多态性位点,因此需要加强保护。2)本研究结果支持仍将紫花苜蓿复合体3个分类群划分为3个种。3)SSR和ISSR标记作为研究紫花苜蓿复合体的遗传多样性及遗传关系是非常有效的分子标记。
致谢:感谢牧草植物分类学专家崔乃然教授的指导,感谢新疆畜牧科学院李捷老师提供的帮助。
[1]秦峰梅,张红香,武祎,等.盐胁迫对黄花苜蓿发芽及幼苗生长的影响[J].草业学报,2010,19(4):71-78.
[2]邹亚丽,韩方虎,耿丽英,等.温度和湿度对紫花苜蓿土壤氮矿化的影响[J].草业学报,2010,19(4):101-107.
[3]王平,周道玮,姜世成.半干旱地区禾-豆混播草地生物固氮作用研究[J].草业学报,2010,19(6):276-280.
[4]Gunn C R,Skrdla W H,Spencer H C.Classification ofMedicagosativaL.using Legume Characters and Flower Colors[M].Washington D C:United States Department of Agriculture Press,1978:84.
[5]中国科学院中国植物志编辑委员会.中国植物志(第42卷第2分册)[M].北京:科学出版社,1998:304-328.
[6]Small E,Jomphe M.A synopsis of the genusMedicago(Leguminosae)[J].Canadian Journal of Botany,1988,67:3260-3294.
[7]Lesins K A,Lesins I.GenusMedicago(Leguminosae):A Taxogenetic study[M].The Hague:W.Junk Press,1979:66-214.
[8]何咏松,吴仁润.苜蓿自交不亲和性的研究[J].草业科学,1987,4(4):6-11.
[9]李拥军,苏加楷.中国苜蓿地方品种亲缘关系的研究Ⅰ种子贮藏蛋白标记[J].草业学报,1999,8(1):31-41.
[10]李拥军,苏加楷.中国苜蓿地方品种亲缘关系的研究ⅡRAPD标记[J].草业学报,1999,8(3):46-53.
[11]王俊杰.中国黄花苜蓿野生种质资源研究[D].内蒙古:内蒙古农业大学,2008.
[12]Diwan N,Bhagwat A A,Bauchan G R,etal.Simple sequence repeat DNA markers in alfalfa and perennial and annualMedicagospecies[J].Genome,1997,40(6):887-895.
[13]Mengoni A,Ruggini C,Vendramin G G,etal.Chloroplast microsatellite variations in tetraploid alfalfa[J].Plant Breeding,2000,119(6):509-512.
[14]Falahati-Anbaran M,Habashi A A,Esfahany M,etal.Population genetic structure based on SSR markers in alfalfa(MedicagosativaL.)from various regions contiguous to the centres of origin of the species[J].Journal of Genetics,2007,86(1):59-63.
[15]Crochemore M L,Huyghe C,Kerlan M C,etal.Partitioning and distribution of RAPD variation in a set of populations of theMedicagosativacomplex[J].Agronomie,1996,16:421-432.
[16]Ghérardi M,Mangin B,Goffinet B,etal.A method to measure genetic distance between allogamous populations of alfalfa(Medicagosativa)using RAPD molecular markers[J].Theoretical and Applied Genetics,1998,96:406-412.
[17]Kidwell K K,Bingham E T,Woodfield D R,etal.Relationships among genetic distance,forage yield and heterozygosity in isogenic diploid and tetraploid alfalfa populations[J].Theoretical and Applied Genetics,1994,89:323-328.
[18]Skinner D Z.Non random chloroplast DNA hyper variability inMedicagosativa[J].Theoretical and Applied Genetics,2000,101:1242-1249.
[19]Havananda T,Brummer E C,Maureira-Butler I J,etal.Relationships among diploid members of theMedicagosativa(Fabaceae)species complex based on chloroplast and mitochondrial DNA sequences[J].Systematic Botany,2010,35(1):140-150.
[20]Bernadette J,Sandrine F,Philippe B,etal.Construction of two genetic linkage maps in cultivated tetraploid alfalfa(Medicagosativa)using microsatellite and AFLP markers[J].Plant Biology,2003,3:9.
[21]Qurios C F,Bauchan G R.Alfalfa and Alfalfa Improvement:The GenusMedicagoand the Origin of theMedicagosativaComplex[M].Madison,Wisconsin:American Society of Agronomy,Crop Science Society of America,Soil Science Society of America Press,1988:93-124.
[22]刘志鹏,杨青川,呼天明,等.用SSR标记研究不同耐盐特性四倍体紫花苜蓿的遗传多样性[J].作物学报,2006,32(4):630-632.
[23]Yeh F C,Yang R-C,Boyle T B J,etal.Popgene:Microsoft Windows?-Based Freeware for Population Genetic Analysis,Version 1.32[M].Canada:University of Alberta and Centre for International Forestry Research,1997.
[24]Peskall R,Smouse P E.GENEALEX 6:Genetic analysis in Excel.Population genetic software for teaching and research[J].Molecular Ecology Notes,2006,6:288-295.
[25]Botstein D,White R L,Skolnick M,etal.Construction of genetic linkage map in man using restriction fragment length polymorphisms[J].American Journal of human genetics,1980,32:314-331.
[26]Takezaki N,Nei M,Tamura K.POPTREE2:Software for constructing population trees from allele frequency data and computing other population statistics with windows interface[J].Molecular Biology and Evolution,2010,27(4):747-752.
[27]Felsenstein J.Confidence limits on phylogenies:an approach using the bootstrap[J].Evolution,1985,39(4):783-791.
[28]Sun G L,Díaz O,Salomon B,etal.Genetic diversity inElymuscaninusas revealed by isozyme,RAPD,and microsatellite markers[J].Genome,1999,42:420-431.
[29]Hilde N,Lgor V B.Effective of life history traits and sampling strategies on genetic diversity estimates obtained with RAPD markers in plants[J].Perspectives in Plant Ecology,Evolution and Systematic,2000,3(2):93-114.
[30]Hamrick J L.Gene Flow and Distribution of Genetic Variation in Plant Populations[M].London:New York Academes Press,1987:53-68.
[31]Wright S.Evolution in Mendelian population[J].Anatomical Record-Advances in Integrative Anatomy and Evolutionary Biology,1929,44:287.
[32]Rieseberg L H,Carney S E.Tansley review no.102plant hybridization[J].New Phytologist,1998,140:599-624.
[33]Gross B L,Kane N C,Lexer C,etal.Reconstructing the origin ofHelianthusdeserticola:survival and selection on the desert floor[J].The American Naturalist,2004,164:145-156.
[34]Brummer E C,Kochert G,Bouton J H.RFLP variation in diploid and tetraploid alfalfa[J].Theoretical and Applied Genetics,1991,83:89-96.
[35]Havananda T,Brummer E C,Maureira-Butler I J,etal.Relationships among diploid members of theMedicagosativa(Fabaceae)species complex based on chloroplast and mitochondrial DNA sequences[J].Systematic Botany,2010,11:140-150.
[36]孙毅,梁爱华,王景雪,等.根据核糖体DNA ITS序列分析苜蓿属的系统分类[J].西北植物学报,2003,23(2):242-246.
猜你喜欢
河北科技师范学院学报(2022年2期)2022-08-26
现代畜牧科技(2021年9期)2021-10-13
现代畜牧科技(2021年4期)2021-07-21
浙江中医药大学学报(2021年6期)2021-07-12
中国粮油学报(2020年12期)2021-01-09
草地学报(2018年5期)2018-11-07
中国病理生理杂志(2018年6期)2018-01-22
中国三峡(2017年4期)2017-06-06
为了孩子(3~7岁)(2016年6期)2016-05-14
原子与分子物理学报(2015年1期)2015-11-24
