
奈帕芬胺合成工艺的改进
2012-05-23 12:12杨巧宾晋继增蒲俊勇李勤耕重庆医科大学药学院重庆400016
中国药房 2012年25期
杨巧宾,晋继增,蒲俊勇,李勤耕(重庆医科大学药学院,重庆 400016)
奈帕芬胺(Nepafenac,A)是由美国Alcon公司研发,2005年8月美国食品与药物管理局(FDA)批准用于治疗与白内障手术相关的疼痛和炎症的一种新型眼用非甾体类解热镇痛抗炎药,与传统的非甾体抗炎药(NSAIDs)相比,具有渗透力强、靶向作用强、毒副作用小等优点[1]。
A的合成方法文献报道主要有2种:(1)N-氯代丁二酰亚胺(NCS)与2-(甲硫基)乙酰胺(D)反应后再与2-氨基二苯甲酮(E)缩合重排,得到中间体2-氨基-3-苯甲酰基-α-(甲硫基)苯乙酰胺(F),再经脱硫得A[2]。(2)E与次氯酸叔丁酯反应后与D缩合重排,得到F,再经脱硫得A[3]。笔者重复方法(1)中制备中间体F时发现反应体系杂质较多,纯化困难,且达不到文献报道[2]的收率(86.4%);而方法(2)中间体F的制备所需温度为-70℃,能源消耗大,还需用到极易发生分解的次氯酸叔丁酯,且该方法收率较低(43%),不适合大规模生产。
本研究参考相关文献[2~5],改变路线(1)中的投料顺序,先将E与NCS氯代,再与D缩合重排,经脱硫即可得A。制备中间体F时反应温度控制在-15℃,条件温和、操作简便、杂质少,收率可达80%以上,只需简单处理则A的纯度可达99.5%以上(高效液相色谱(HPLC)归一化法)。本工艺总收率可达48.6%(各步反应收率之乘积:86%×78.2%×88%×82.2%),适合大规模生产,笔者在此将该合成方法介绍如下。
1 仪器与试药
1.1 仪器
JT302 N型电子天平(上海精天电子仪器有限公司);YRT-3型熔点仪(南京旭析仪器有限公司);KF-8 A卡尔费休水分仪(上海佳实电子科技有限公司);1200 HPLC仪(美国安捷伦公司);Avance 400核磁共振仪(瑞士Bruker公司)。
1.2 试药
金属钠(批号:20110409,纯度:99.7%)、巯基乙酸甲酯(B,批号:20110406,纯度:96%)、2-氨基二苯甲酮(E,批号:20110512,纯度:98%)、NCS(批号:20110311,纯度:98%)、雷尼镍催化剂均由上海晶纯实业有限公司提供;硫酸二甲酯(成都西亚试剂有限公司,批号:20110315,纯度:99.5%);氨水、三乙胺(Et3N)、甲醇、二氯甲烷、无水硫酸钠、异丙醚、四氢呋喃、异丙醇等均为分析纯。
2 方法与结果
2.1 合成路线
奈帕芬胺的合成路线如图1所示。
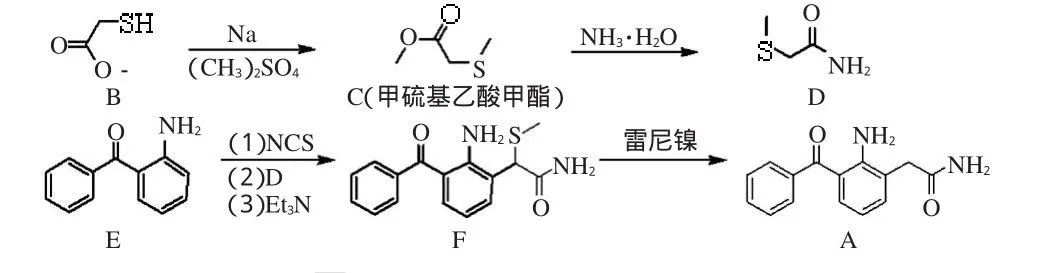
图1 奈帕芬胺合成路线Fig 1 Synthetic route of nepafenac
2.2 合成方法
2.2.1 甲硫基乙酸甲酯(C)的合成。取金属钠(8.2g,0.356 mol)分批加至冰浴的甲醇(260mL)中,降温至-10℃,氮气保护下滴加巯基乙酸甲酯(B,31.9mL,0.356 mol),加毕搅拌10min,缓慢滴加硫酸二甲酯((CH3)2SO4,33.7mL,0.356 mol),加毕升至室温反应4h。减压浓缩至干,剩余物中加入水(100mL)、二氯甲烷(200mL),搅拌10min,水洗二氯甲烷层至中性,用无水硫酸钠干燥后过滤,滤液减压浓缩,得淡黄色液体C(36.7 g,收率:86%,文献[6]收率:78.4%)直接用于下步反应。
2.2.2 2-(甲硫基)乙酰胺(D)的合成。取C(36.7 g,0.306 mol)置于圆底烧瓶中,冰浴滴加氨水(84.9 g,94.3mL),加毕升至室温搅拌1h,降温至0~5℃,搅拌2h,过滤,滤饼用异丙醇(20mL)润洗,干燥得白色固体粉末(25.1 g,收率:78.2%,熔点(mp):99~102℃ ;文献[2]收率:76.2%,mp:99~102℃,纯度:98.2%(HPLC归一化法))。
2.2.3 2-氨基-3-苯甲酰基-α-(甲硫基)苯乙酰胺(F)的合成。取E(28.7 g,0.146 mol)加入氢化钙(CaH)干燥过的二氯甲烷400mL,搅拌溶解后降温至-15℃,氮气保护下将NCS(19.5 g,0.146 mol)溶于200mL二氯甲烷缓慢滴加至反应液中,加毕搅拌0.5 h,将D(15.3 g,0.146 mol)溶于200mL二氯甲烷并缓慢滴加至反应液,-15℃保持3 h后缓慢升温至0~8℃。立即过滤反应液,将得到的滤饼倒至二氯甲烷(500mL)中,冰浴滴加三乙胺(20mL)至二氯甲烷中,搅拌15min,水洗二氯甲烷层至中性,无水硫酸钠干燥后过滤,滤液减压浓缩,得黄色固体粉末。加入异丙醚(500mL),搅拌回流1 h,降至室温,过滤,干燥得黄色固体(38.5 g,收率:88%,mp:153~154.5℃;文献[3]收率:43%,mp:153~155℃,纯度:99.5%(HPLC归一化法))。
2.2.4 奈帕芬胺(A)的合成。取F(15 g,0.05 mol),加入四氢呋喃(300mL)搅拌溶清,称取雷尼镍(60 g)以水洗3次、四氢呋喃洗3次后快速加至反应液中,搅拌15min,过滤。滤饼用四氢呋喃(100mL)润洗,滤液减压浓缩,得黄色固体(13 g),用异丙醇重结晶,得黄色针状结晶A(10.4 g,收率:82.2%,mp:178.5~180℃;文献[3]收率:73%,mp:178~180℃,纯度:99.7%(HPLC归一化法))。
2.3 目标化合物的结构分析
对A进行结构分析,得到以下数据:mp:178.5~180℃,1H-NMR(400MHz,CDCl3):δ 7.52~7.56(m,5 H),7.07(s,2 H),7.30~7.31(d,1 H),7.20~7.22(d,1 H),6.53~6.57(t,1 H),3.41(s,2 H),3.33(s,2 H)。结果与文献[3]一致,由此可知所得产物为奈帕芬胺。
2.4 反应条件的筛选
本文主要考察了制备关键中间体F的反应条件。
2.4.1 反应温度的影响。文献[3]报道反应温度在-70℃,此温度下工业生产所需能源消耗大、成本高、收率低;升高反应温度至-40℃,采用NCS来作氯代原料,然而此方法的投料顺序产生杂质多,难以纯化。在本试验中,操作条件同“2.2.3”项下方法所述,笔者考察了-65℃、-45℃、-15℃、0℃不同反应温度下,反应时间2~5 h时的收率,结果见表1。
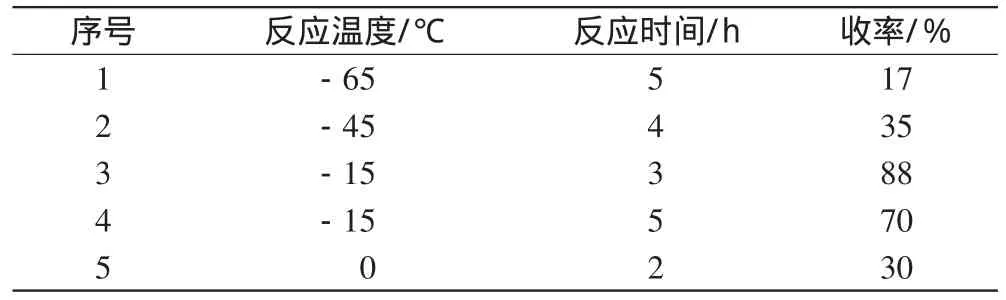
表1 反应温度和反应时间对收率的影响Tab 1 Effect of reaction temperature and reaction time on yield
由表1可见,反应温度在-15℃时为宜。低于-15℃时,反应时间需要延长;高于-15℃时,收率降低,并且副产物增多;在-15℃反应温度下反应3 h就可以达到令人满意的收率,延长反应时间至5 h,收率有所下降。故最终选择-15℃反应3 h为合适的反应条件。
2.4.2 二氯甲烷中的水分对反应的影响。在制备中间体F时,笔者发现溶剂中的水分对产物的纯度和收率有很大的影响,由此考察了二氯甲烷的水分对收率的影响,结果见表2。

表2 二氯甲烷中的水分对收率的影响Tab 2 Effect of the moisture content of methylene chloride on yield
由表2可见,二氯甲烷中的水分应该严格控制在0.15%以下,由于工业生产中工业级二氯甲烷水分一般高于0.3%,本试验虽然用的是分析纯二氯甲烷,但是经检测其水分均高于0.3%。因此在制备中间体F时对二氯甲烷水分用卡尔费休水分测定仪先进行水分检测。一般实验室中,二氯甲烷的水分可用CaH或者五氧化二磷回流干燥蒸馏方法去除;生产中可以用分子筛干燥去除。
3 讨论
在制备中间体F时,二氯甲烷对D和NCS的溶解度较差,笔者曾尝试用四氢呋喃代替二氯甲烷作溶剂,但由于四氢呋喃易吸潮,结果并未提高产率,因此只能通过增加二氯甲烷的用量来提高对反应底物的溶解度。
制备中间体F时,-15℃可以用冰盐浴来控制反应温度;在控制二氯甲烷中的水分时,笔者尝试用氯化钙来干燥去除,结果发现干燥1 d后,水分低于0.15%,可达到反应要求,因此工业生产时可用氯化钙来干燥二氯甲烷。
本文解决了奈帕芬胺生产时所需苛刻条件,降低了工业生产成本,为奈帕芬胺滴眼液的制备提供了高纯度的原料。
本试验结果表明,本方法所得收率与文献比较更高,并且反应条件温和,达到了良好的效果,是一条适用于工业化生产奈帕芬胺的合成路线。
[1]刘 岩,王林洪,张 潇.奈帕芬胺在眼科应用的研究进展[J].中国实用眼科杂志,2010,28(10):1060.
[2]Suarez GT,Escude GA,Valles SC.Process for preparing a benzoylbenzeneacetamide derivative[P].US:20090312575,2009-12-17.
[3]Walsh DA,Wayne WH,Shamblee DA,et al.Syntheses and biological evaluation of potential prodrugs of 2-amino-3-benzoylbenzeneacetic acid and 2-amino-3-(4-chlorobenzoyl)benzeneacetic acid[J].J Med Chem,1990,33(8):2296.
[4]Gassman PG,Van Bergen TJ.Oxindoles,a new,general method of synthesis[J].J Am Chem Soc,1974,96(17):5508.
[5]Gassman PG,Gruetzmacher G,Van Bergen TJ.Generation of azasulfonium salts from halogen-sulfide complexes and anilines,the synthesis of indoles,oxindoles,and alkylated aromatic amines bearing cation stabilizing substituents[J].JACS,1974,96(17):5512.
[6]Carpenter JM,Shaw G.Synthesis of 5-methylsulphonyluridine and its 5′-mono-and pyro-phosphates[J].J Chem Soc C,1970,15:2016.
猜你喜欢
化工设计(2022年4期)2023-01-02
贵州科学(2022年4期)2022-09-05
中国药学药品知识仓库(2022年10期)2022-05-29
化学工程师(2022年3期)2022-04-19
汕头大学学报(自然科学版)(2020年4期)2020-12-14
商情(2020年17期)2020-11-28
现代盐化工(2019年4期)2019-09-10
中成药(2017年5期)2017-06-13
科技与创新(2015年20期)2015-10-29
科技与企业(2015年20期)2015-10-21
