
基于新型交联聚合物电解质的准固态染料敏化太阳能电池
2012-03-06 04:43梁桂杰钟志成张增常陈美华李在房候秋飞
物理化学学报 2012年12期
梁桂杰 钟志成 许 杰 张增常 陈美华李在房 和 平 候秋飞
(1湖北文理学院低维光电材料与器件湖北省重点实验室,湖北襄阳441053;2西安交通大学金属材料强度国家重点实验室,西安710049;3武汉纺织大学新型纺织材料绿色加工及其功能化教育部重点实验室,武汉430073)
1 引言
自从1991年,Oregan和Grätzel教授1在Nature上报道了染料敏化太阳能电池(DSSC)以来,DSSC以其高效、工艺简单、成本低廉等系列优势在全世界范围内引起了广泛关注,并成为新一代光伏电池的候选物得到大力发展.如今,基于有机液态电解质的DSSC的光电转化效率达到12.3%,2已接近目前商业化非晶硅太阳能电池的水平.但是,通常液态电解质易挥发、泄漏,造成电池封装困难,电池的长期稳定性下降.随后,各种固态或准固态电解质如:有机/无机空穴传输材料、3-5电子导电聚合物、6-8无机p型半导体9-12和聚合物电解质13-16等作为液态电解质的替代品应运而生.聚合物电解质一般是由氧化还原性的导电离子对(I-/I-3)溶于半晶态的聚合物之中形成,聚合物基体作为骨架结构为导电离子的传输提供通道,导电离子被包覆在聚合物基体的三维网络结构之中形成稳定的电解质体系.由于蒸汽压低,可塑性强,并且对多孔光阳极的渗透性好,聚合物电解质不仅可以解决常规液态电解质的挥发强、电池使用寿命短的问题,而且可以克服一般固体电解质与电极之间界面接触差、电池效率低的缺陷,因而一度成为DSSC用电解质体系中研究的热点.
在众多的聚合物电解质中,聚氧化乙烯(PEO)基聚合物电解质,因其基体材料的分子结构柔顺、熔点较低、成膜性好且对碱金属盐的溶解性能好等特点,17使其成为性能优异的电解质材料.然而,PEO高聚物自身较大的结晶度使得其聚合物电解质的室温离子电导率较低(10-7-10-6S·cm-1),18电池光电性能受到限制.为此,研究者们采用聚合物共混、19,20交联、21,22添加增塑剂、12,23无机粒子填充24-26等一系列的方法对PEO基聚合物电解质进行改性,并且获得了较好的效果.本文采用低分子量的聚乙二醇(oligo-PEG)代替常规的PEO高聚物,选用柠檬酸(CA)作为交联剂,合成聚柠檬酸-乙二醇(PCE)基体材料,并将PCE聚酯基体浸入到含有LiI、I2的电解液中,得到具有一定液含量的准固态三维交联型PCE/LiI/I2聚合物电解质.该电解质与常规PEO基电解质相比,一方面,oligo-PEG的引入在保留了-C-C-O-链段(PEO的主链)良好的柔顺性和对导电盐溶解性的同时,有效降低了聚合物基体的结晶度,增强了电解质对多孔光阳极的渗透性,使得聚合物电解质的电导率增大,并且基体分子间化学交联作用的存在使电解质的稳定性提升;另一方面,PCE聚酯基体材料良好的生物相容性和生物降解性(研究表明其试样在磷酸盐缓冲溶液中浸泡65 h后降解失重都超过50%27)将赋予PCE/LiI/I2电解质优良的可生物降解性,这将有效地减轻目前大量废弃的聚合物电解质对环境造成的危害.
本文旨在制备一种可生物降解的、交联网络型准固态聚合物电解质,使其兼有固态电解质的稳定性和液态电解质较高的导电性,以期得到高效、稳定且清洁环保的DSSC.文中,从分子结构着手,首先研究了PEG的分子量对PCE交联聚酯基体的微观形貌及吸液性能的影响,并探讨了PCE基体的吸液机理;接着以此为基础,进一步研究了PEG的分子量对PCE/LiI/I2电解质导电性能及其准固态DSSC光电性能的影响.
2 实验部分
2.1 PCE/LiI/I2交联聚合物电解质的制备

图1 PCE/LiI/I2交联聚合物电解质的形成示意图Fig.1 Formation of the PCE/LiI/I2crosslinked polymer electrolyte
实验所用药品试剂除特别说明外,均为分析纯的国药集团化学试剂有限公司产品,使用前所有药品和试剂都进行干燥脱水处理.PCE/LiI/I2交联聚合物电解质的形成包括两大步骤,其形成过程如图1所示.步骤1为PCE交联聚合物基体的合成:将摩尔比为2:3的PEG(Mˉw=200,400,1000,2000)和CA依次加入到盛有一定量二甲苯溶剂的三口烧瓶中,三口烧瓶上加置油水分离器、电动搅拌器和温度计.反应过程中向反应容器中通入N2加以保护,PCE交联聚合物基体的具体反应过程及条件见文献.28步骤2为液态电解质的吸收及聚合物电解质的成型:先将LiI/I2按照0.3 mol·L-1/0.03 mol·L-1的摩尔浓度分别溶于乙腈(ACN)、N-甲基吡咯烷酮(NMP)和丙烯碳酸酯(PC)中,配制成三种液态电解质溶液;将制备的PCE交联聚合物基体膜放入上述配制的电解液中浸渍24-48 h,然后取出,用滤纸拭去其表面残留的溶液至质量恒重,得到准固态的PCE/LiI/I2交联聚合物电解质.
2.2 DSSC的组装
TiO2光阳极及铂对电极的制备参照文献.29将上述制备的PCE/LiI/I2聚合物电解质升温至70°C(不超过电解液溶剂的沸点),提高聚合物电解质的流动性;然后将其涂布在经N3染料(cis-bis(isothiocyanato)bis(2,2ʹ-bipyridyl-4,4ʹ-dicarboxylate)ruthenium (II),澳大利亚Dyesol公司)敏化后的TiO2光阳极薄膜上;接着将铂对电极覆盖在聚合物电解质表面上,用夹子将两侧电极夹紧,放置约30 min至聚合物电解质降至室温,这样使高温聚合物电解质在两侧电极的夹持力下充分渗透到TiO2多孔薄膜中,并在冷却至室温的过程中固化成型;将TiO2光阳极、聚合物电解质和铂对电极按顺序组装成“三明治”式结构,四周用热封薄膜(Surlyn1702,大连七色光太阳能科技开发有限公司)封装,从而组装得到准固态染料敏化太阳能电池.
2.3 性能测试与表征
2.3.1 PCE交联聚合物基体的分子结构表征
采用傅里叶变换红外(FTIR)光谱仪(VERTEX70,德国Buber公司),选用KBr压片法,并结合透射方式对样品进行红外测试,扫描波数范围为400-4000 cm-1,扫描速率为2 cm-1·s-1,测试前样品在红外灯照下干燥除水处理.
以二甲苯为溶剂,将交联聚合物产物置于索氏抽提器中抽提过滤48 h,取出在40°C的真空烘箱中干燥48 h;取少量上述样品放入氘代二甲基亚砜(DMSO,上海柏卡化学技术有限公司)试剂中,加热并超声分散,待样品溶解后放入核磁共振仪(AV400,瑞士Bruker公司)中进行1H-NMR测试.
2.3.2 聚合物电解质基体膜的微观形貌表征
将制备的PCE聚合物电解质置于冷冻干燥机(VFD-1000,北京博医康实验仪器有限公司)中48 h,至电解液升华完全后得到聚合物电解质基体的冻干膜.将该膜经液氮脆断后,采用扫描电子显微镜(SEM,HITACHI X-650,Netherlands FEI corp.)观察膜的断面形貌,测试加速电压为10 kV,测试前样品经过干燥和蒸镀喷金处理.
2.3.3 聚合物电解质基体膜的孔隙率测定
将上述制备的聚合物电解质基体的冻干膜放入45°C的真空烘箱中,干燥24 h,取出放于干燥器中待用.在聚合物电解质基体膜上截取直径为1 cm的均匀圆片,将称好质量(mdry)和测好厚度的圆片基体膜(表观体积为V)在去离子水中浸泡2 h后取出,用滤纸吸干膜表面的水分后称重(mwet),根据膜浸泡前后的质量变化计算膜的孔隙率(P):30
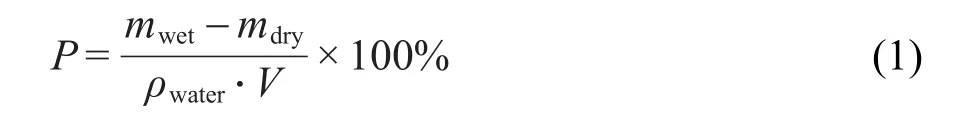
2.3.4 聚合物电解质的吸液性能测试
将PCE聚酯基体膜在40°C的真空烘箱中干燥至质量恒定,取质量为m0(单位g)的干态PCE样品膜放入带塞子的磨口瓶中,在含有0.3 mol·L-1/0.03 mol·L-1的LiI/I2的电解质溶液中浸泡,间隔一定时间取出试样,用滤纸拭去其表面多余的溶液至质量恒定,得到质量为m的PCE凝胶,吸液率计算如下:

式中,Qle为PCE基体对液态电解质的吸液率(g· g-1);m为达到吸液平衡时PCE凝胶的质量(g).
2.3.5 聚合物电解质的拉曼光谱测试
将PCE/LiI/I2电解质样品在40°C的真空恒温干燥箱中干燥48 h,备用.在室温下,采用拉曼光谱仪(FRA 106/s,德国Buber公司)对样品膜进行拉曼光谱测试.样品扫描波数范围为50-200 cm-1,扫描分辨率为1 cm-1.
2.3.6 聚合物电解质的离子导电性能测试
PCE/LiI/I2电解质中扩散系数的测试:用化学电镀方法在透明导电玻璃FTO上镀一层厚度约为2 μm的铂层作为铂电极,选用两块对称的铂片电极,在其间夹置电解质,组装成结构为Pt/(PCE/LiI/ I2)/Pt的化学电池,电解质膜的面积为1 cm×1 cm,厚度约为50 μm.室温下,用电化学工作站(LK9805,天津兰力科化学电子高技术有限公司)选用线性扫描伏安法测试Pt/(PCE/LiI/I2)/Pt化学电池的I-V曲线,电位扫描范围为-1.0-1.0 V,电位扫描速率为10 mV·s-1.
PCE/LiI/I2电解质离子电导率(σ)的测试与计算方法参照文献.31
2.3.7 DSSC的光电性能测试
模拟太阳光光源为上海蓝晟电子有限公司生成的500 W氙灯,用北京京合绿能太阳能公司生产的阳光辐照计YFJ10来控制模拟光的入射强度.在入射光强为60 mW·cm-2下,采用LK9805型电化学工作站,选用线性扫描法测试DSSC的I-V曲线,电池有效面积为0.5 cm2,电位扫描范围为0-0.8 V,扫描速率为0.01 V·s-1.
3 结果与讨论
3.1 PCE交联聚合物基体的分子结构分析
CA与PEG通过酯化反应能够生成稳定的PCE交联聚合物网络,PCE交联聚合物基体的IR光谱图见图2.2872 cm-1处附近对应的是PEG分子上-CH2-的伸缩振动吸收峰,在PCE交联产物中该处的峰强随着PEG分子量的增大而增大,这是因为当参加反应的PEG的摩尔量一定时,随着分子量的增加,PEG分子中-CH2-单元数逐渐增加;根据文献,271732 cm-1处的较强的吸收峰为酯羰基碳氧双键的伸缩振动吸收峰,这应归属为PEG与CA之间通过酯化反应生成的酯羰基的吸收峰;在纯PEG中,1104 cm-1处附近存在较强的-OH的弯曲振动吸收峰,在PCE交联产物中其值明显减小,说明PEG的端羟基与CA的羧基之间发生了酯化反应.

图2 PCE交联聚合物基体的IR光谱图Fig.2 IR spectra of the PCE crosslinked polymer matrices
为了进一步弄清PCE交联产物的分子结构,以CA-PEG400为例对其进行了1H-NMR测试,结果见图3.样品在化学位移(δ)为4.30和4.11处出现面积较大的多重峰,为PEG400分子中-CH2-单元的质子共振吸收峰;δ=3.77处出现一个小面积的单峰,为CA分子上-C-OH单元的质子共振吸收峰;δ=3.52处出现的多重峰,为PEG400分子中-OCH2CH2O-单元的共振吸收峰;δ=2.86和δ= 2.78处出现的多重峰,为CA分子上-CH2-单元的共振吸收峰;δ=2.51处为DMSO溶剂的共振吸收峰.图3显示,δ=3.77(CA分子上的-C-OH单元)和δ= 3.52(PEG400分子中-OCH2CH2O-单元)处两个吸收峰的峰面积分别为4.291和45.004,即CA分子上的-C-OH单元和PEG400分子中-OCH2CH2O-单元的个数比为1:10.4,将其转换成CA和PEG400的分子个数比为1:1.55,这与图1中所示的交联产物结构式中CA与PEG400之间的比例2:3基本接近.由此,PCE交联产物的分子结构得以验证.
3.2 PEG分子量对聚合物电解质基体膜微观结构的影响
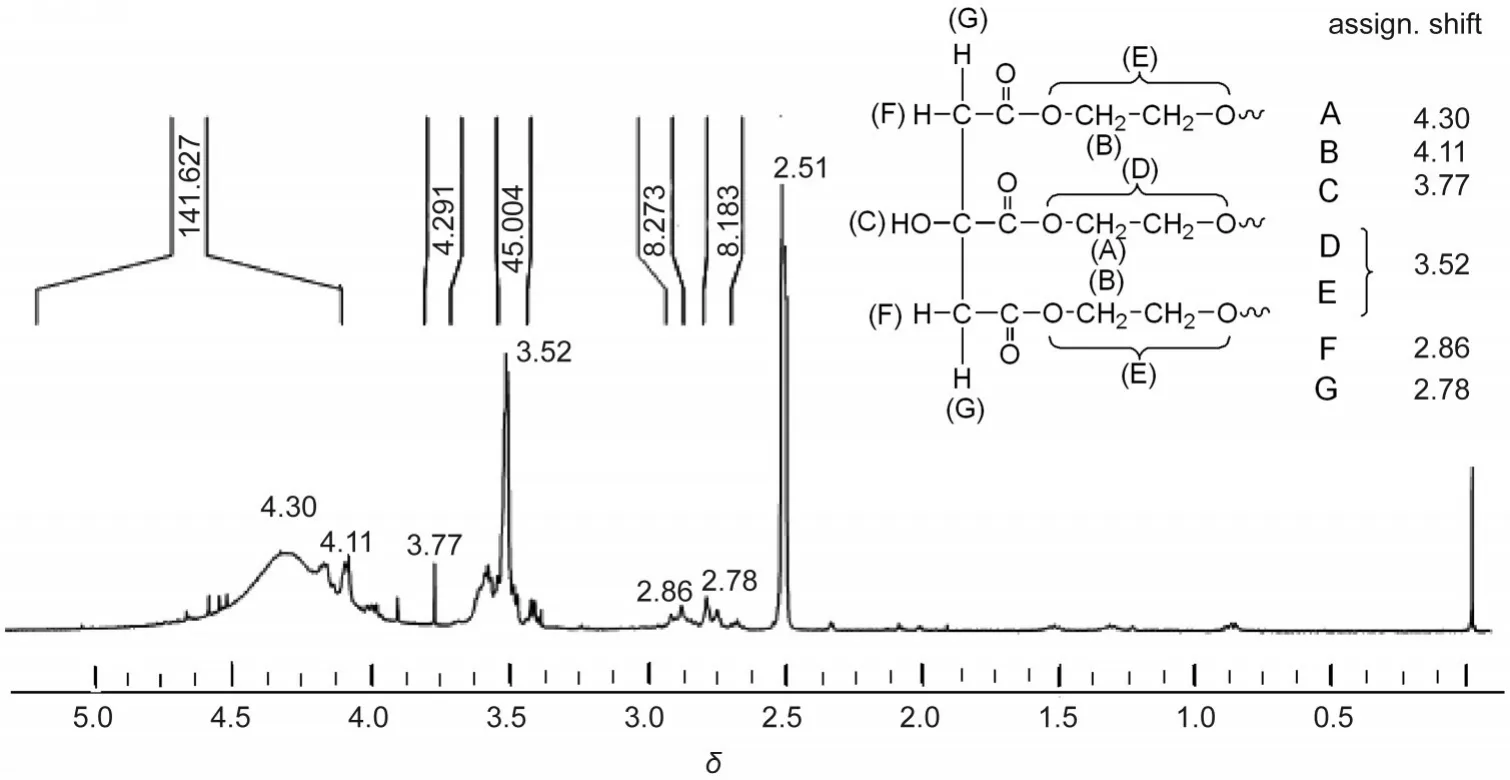
图3 CA-PEG400交联聚合物基体的1H-NMR图Fig.3 1H-NMR spectrum of the CA-PEG400 crosslinked polymer matrix

图4 聚合物电解质冻干膜的截面SEM图Fig.4 SEM images of the cross section of the freeze-drying polymer electrolyte membranes(a)CA-PEG200/LiI/I2,(b)CA-PEG400/LiI/I2,(c)CA-PEG1000/LiI/I2,(d)CA-PEG2000/LiI/I2
PCE交联基体吸收电解液后成为聚合物电解质.将聚合物电解质置于冻干机中48 h,至电解液升华完全后得到冻干膜,图4为聚合物电解质冻干膜的截面SEM图.当PEG分子量Mˉw=200时,膜的表面较为平滑(除去成膜过程中外因造成的裂痕),整个膜显得较为密实,这可能是因为PEG200的分子链段短,加之其单位质量内的端羟基数目多,交联程度大,导致膜的结构较为紧密;PEG分子量为400时,随着PEG分子量的增大,聚合物基体膜的结构开始变得疏松;当PEG分子量达到1000和2000时,膜的表面明显变得粗糙,膜的结构更加疏松.由此可见,PEG的分子量影响聚合物电解质基体膜的微观结构(疏松程度).为了定量地描述其影响效果,接下来我们测定了基体膜的孔隙率.由表1中孔隙率的测试结果可知,改变PEG的分子量可以控制聚合物电解质基体膜的孔隙率,基体膜的孔隙率随着PEG分子量的增加(Mˉw=200,400,1000,2000)而逐渐增大.
3.3 PEG分子量对聚合物电解质吸液性能的影响
PEG分子量决定着聚合物基体的微观结构(见3.2节分析),从而影响聚合物基体对电解液的吸收性能.图5是含有不同分子量PEG的聚合物基体的吸液率图,对于三种不同的电解液(ACN/LiI/I2、PC/ LiI/I2和NMP/LiI/I2),聚合物基体的吸液率均随着PEG分子量的增加而增大.该变化趋势可以由聚合物基体的微观形貌随PEG分子量的变化及由其导致的基体吸液机理的变化来做解释:当PEG的分子量Mˉw=200,400时,由于聚合物基体较为密实,使其主要依靠基体与电解液之间的酸碱作用来吸收液态电解质.另外,由于低分子量的PEG200和PEG400在单位质量内含有较多的端羟基可以与CA上的羧基发生交联反应,PEG与CA之间较强的氢键作用力不利于基体中的PEG组分的溶胀,以上原因导致其对电解液的吸收效率较低;当PEG的分子量增大到1000时,聚合物基体的孔隙率明显增加,电解液可以直接渗透到基体的孔隙内部,此时,基体对电解液的吸收不仅依靠两者之间的酸碱作用,而且还依赖于基体的三维网络结构对电解液的吸纳与包敷作用,这使得基体的吸液率显著升高.当PEG的分子量达到2000时,聚合物基体的结构更加疏松,其吸液率更高.从图5还可以看出,聚合物基体的吸液率随着溶剂物质的不同而发生变化,吸液率:ACN<PC<NMP.这是因为在一定程度上CA-PEG基体依靠着与电解液之间的酸碱作用来实现其对电解液的吸收过程,溶剂的电子施主数越大其碱性越强,此时,溶剂与酸性的CA-PEG基体之间的Lewis作用力越大,溶剂对基体的渗透溶胀效果越好.ACN、PC和NMP的电子施主数依次增大,因此,聚合物基体对其的吸液率呈现依次升高趋势.

表1 聚合物电解质基体膜的孔隙率Table 1 Porosity of the matrix membranes of the polymer electrolytes

图5 室温(25°C)下不同PCE基体对液态电解质的吸液率Fig.5 Liquid electrolyte uptake of the PCE matrices at room-temperature(25°C)
图6是PCE交联基体对NMP/LiI/I2电解液的吸液率随时间的变化关系曲线,四种交联基体达到各自吸液平衡的时间分别约为60、55、36和25 h,即PCE交联基体对NMP/LiI/I2电解液的吸液速率随PEG分子量的增加而逐渐加快.这是由于较高分子量PEG对应的PCE基体膜的疏松程度较大,从而有效地提高了基体对电解液的吸收速率(原因与吸液率随PEG分子量变化的原因相一致,具体分析同上).以上分析表明,PCE基体膜的微观结构对聚合物电解质的吸液性能起重要作用.
3.4 PEG分子量对聚合物电解质中扩散性能影响
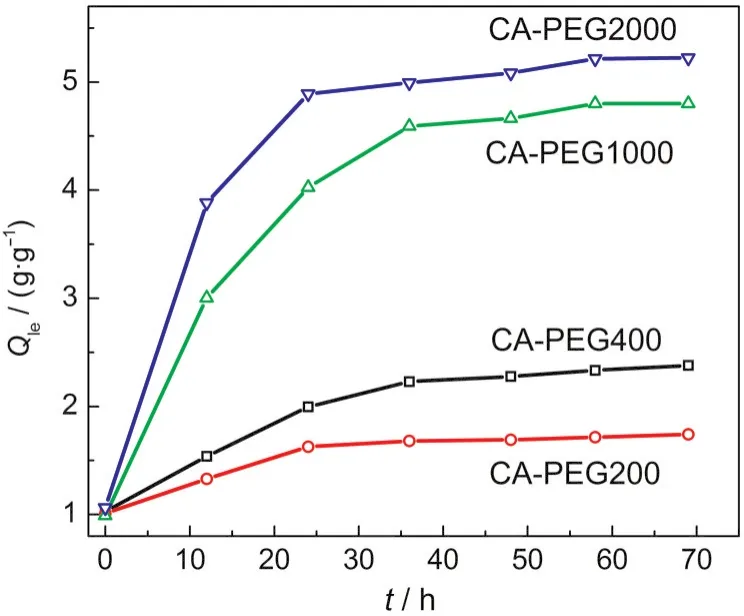
图6 室温(25°C)下PCE基体的吸液率随时间的变化曲线Fig.6 Plots of the liquid uptake of PCE matrices versus the time at room-temperature(25°C)
图7左上角插图为聚合物电解质的Raman光谱图,四种电解质样品在波数为113和141 cm-1处均分别出现了一个较强的吸收峰和一个相对较弱的吸收峰,其分别对应着和高聚碘(如)的对称伸缩振动;32而在波数为180-200 cm-1处没有出现I2的特征振动峰,这说明电解质中的I2已经完全转换成了和.32113 cm-1处的峰强度远比141 cm-1处的强,表明电解质中导电离子的主要存在形式为.随着PEG分子量的增加,113 cm-1处峰的强度逐渐增大,即聚合物电解质中的量随着PEG分子量的增加而增大,这可以用聚合物基体膜吸液率随PEG分子量的变化现象来解释.由于电极界面处离子的氧化还原过程较快,电解质的导电性能由电解质中离子的扩散过程决定.从图7中的伏安曲线可以得出I-3的极限电流密度,的扩散系数值可由公式(3)得出.该实验中由于聚合物电解质中的直接摩尔浓度不便于确定(的摩尔浓度与对应的Raman峰的强度成正比),无法得到的绝对扩散系数值,这里以CA-PEG200//LiI/I2为参照标准,得到四种电解质中I-3的相对扩散系数值(Drelative),结果列于表2. PEG分子量=200,400,1000,2000的聚合物电解质中的相对扩散系数分别为1.00×10-6、1.22×10-6、1.91×10-6和2.62×10-6cm2·s-1,PEG分子量的增加使得电解质中的扩散系数逐渐增大,这可能是因为随着PEG分子量的增加,基体对电解液的吸液率增大而使其不断膨胀,基体内部组分间的交联作用或氢键作用减弱,传输过程中受到的阻力减小,离子扩散更加容易.
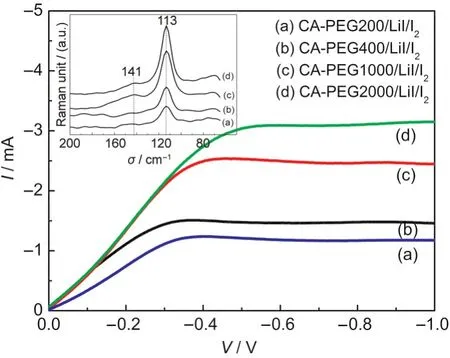
图7 PCE/LiI/I2聚合物电解质的Raman光谱及其对应的电解质的伏安曲线Fig.7 Raman spectra and the corresponding voltammetry curves of the PCE/LiI/I2polymer electrolytes
表2 PCE/LiI/I2聚合物电解质中的相对扩散系数及跃迁活化能值Table 2 Relative diffusion coefficients and transition activation energies ofin the PCE/LiI/I2polymer electrolytes at room temperature

表2 PCE/LiI/I2聚合物电解质中的相对扩散系数及跃迁活化能值Table 2 Relative diffusion coefficients and transition activation energies ofin the PCE/LiI/I2polymer electrolytes at room temperature
y=K·x+m:the fitting straight line equation of the lnσ-1/T plot;K:the slope of the fitting straight line,the value is defined as-Ea/R.R2:the adjusted R2for the fitting straight line equation.Drelative:the relative diffusion coefficient ofin the polymer electrolyte with CA-PEG200/ LiI/I2as reference standard
?

式中,D为导电离子的表观扩散系数(cm2·s-1);Jlim为极限电流密度(mA·cm-2);d为电池两极之间的间距(μm);n为每摩尔分子(I2)发生电化学反应时转移的电子数;F为法拉第常数(C·mol-1);C表示电活性物种()的初始摩尔浓度(mol·L-1).此实验中d=50 μm,n=2;F=96487 C·mol-1.
3.5 PEG分子量对聚合物电解质离子导电性能的影响
图8是聚合物电解质的离子电导率随温度的变化关系图,在25-85°C温度范围内,随着温度的升高,四种聚合物电解质的离子电导率值均逐渐增大.“lnσ-1/T”图形均呈直线关系,说明在实验的温度变化范围内,考虑到实验条件和实验误差的情况下,该聚合物电解质的电导率与温度的关系满足Arrhenius方程:

式中,σ为电解质的离子电导率(S·cm-1);A为指前因子;R为气体常数,其值为8.314 J·mol-1·K-1;T为样品所处的温度(K);Ea为电解质中导电离子的跃迁活化能(kJ·mol-1).
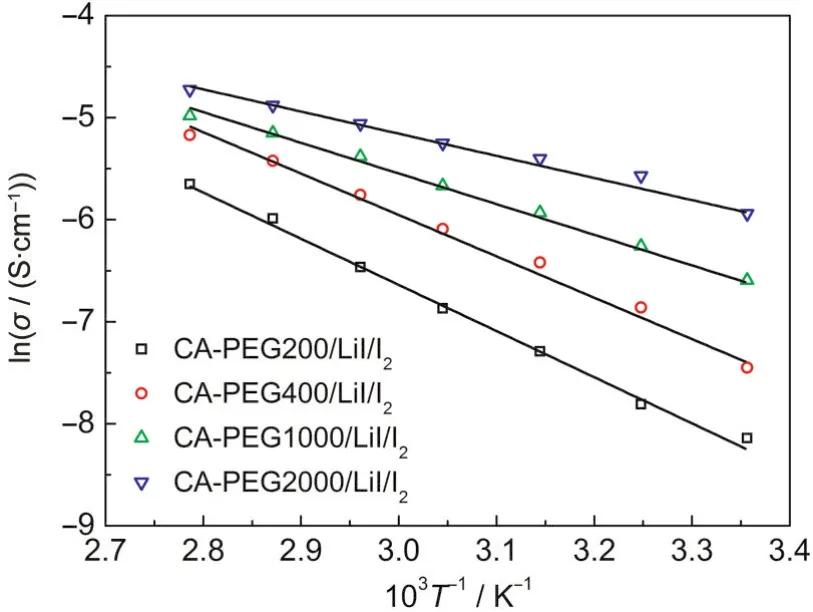
图8 PCE/LiI/I2聚合物电解质的lnσ与1/T之间的关系图Fig.8 Plots of lnσ versus 1/T of the PCE/LiI/I2polymer electrolytes
根据拟合直线的斜率(K=-Ea/R)可以得出电解质中的跃迁活化能(Ea),结果见表2.的跃迁活化能随着PEG分子量的增加而逐渐减小,这可能是因为较大分子量PEG对应的聚合物基体的网络结构更加疏松,基体吸收的电解液较多,自由体积较大,在基体中的扩散更加容易,跃迁活化能降低.
3.6 PEG分子量对DSSC光电性能的影响
将制备的PCE/LiI/I2聚合物电解质用于染料敏化太阳电池中,采用线性扫描伏安法测试了电池的光电流密度-光电压(J-V)曲线,结果如图9所示.从表3中的DSSC的光电性能参数可以看出,随着PEG分子量的增加,聚合物电解质对应的电池的短路光电流密度值依次增加;电池的开路光电压值随PEG分子量的增加变化不明显;PEG分子量Mˉw= 200,400,1000,2000的四种电解质样品对应的电池的光电转化效率(η)分别为3.26%、3.34%、4.26%和4.89%.这可能是因为随着PEG分子量的增加,聚合物基体的网络结构变得疏松,电解质膜的吸液率增加,聚合物电解质膜的离子电导率增大,导致DSSC的光电转化性能提高.
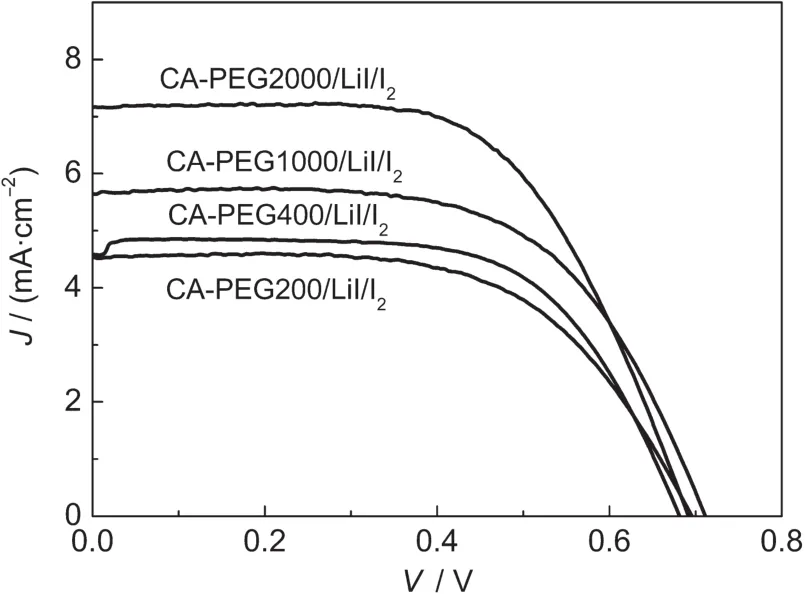
图9 基于PCE/LiI/I2聚合物电解质的DSSC的光电流密度-光电压(J-V)曲线Fig.9 Photocurrent density-photovoltage(J-V)curves of the DSSC based on the PCE/LiI/I2polymer electrolytes
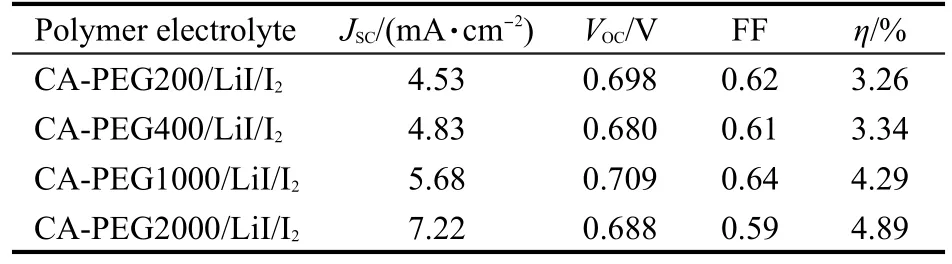
表3 DSSC的光电性能参数Table 3 Photoelectric performance parameters of the DSSC
4 结论
选用短链段的聚乙二醇代替常规的PEO高聚物,选用柠檬酸作为交联剂,合成聚柠檬酸-乙二醇交联聚酯,并将PCE基体浸入到含有LiI、I2的电解液中,制备得到准固态的三维交联型PCE/LiI/I2聚合物电解质.首先采用IR、1H-NMR和SEM表征了PCE基体的分子结构和聚合物电解质的微观形貌,研究了PEG分子量对聚合物电解质微观形貌的影响,结果表明,随着PEG分子量的增加,电解质基体膜的孔隙率逐渐增大,基体膜的疏松度逐渐增加. PCE基体的吸液曲线和聚合物电解质的线性扫描伏安曲线结果显示,PEG分子量Mˉw=200,400,1000, 2000的四种PCE基体的吸液率和吸液速率逐渐增大,其对应的准固态聚合物电解质中I-3的相对扩散系数值(以CA-PEG200//LiI/I2为参照标准)分别为1.00×10-6、1.22×10-6、1.91×10-6、2.62×10-6cm2·s-1,I-3的跃迁活化能值分别为37.60、33.72、24.97和18.04 kJ·mol-1;在25-85°C实验温度范围内,PCE/LiI/I2聚合物电解质的离子电导率随温度的升高而增大,其二者之间关系满足Arrhenius方程.DSSCs的光电性能参数表明,随着PEG分子量Mˉw从200、400、1000增大到2000,对应电池的短路光电流密度依次增大,在60 mW·cm-2的入射光强下,四种电解质对应电池的光电转化效率分别为3.26%、3.34%、4.26%和4.89%.
(1) Oregan,B.;Grätzel,M.Nature 1991,353,737.doi:10.1038/ 353737a0
(2)Yella,A.;Lee,H.W.;Tsao,H.N.;Yi,C.Y.;Chandiran,A.K.; Nazeeruddin,M.K.;Diau,E.W.G.;Yeh,C.Y.;Zakeeruddin,S. M.;Grätzel,M.Science 2011,334,629.doi:10.1126/ science.1209688
(3) Xia,J.;Masaki,N.;Lira-Cantu,M.;Kim,Y.;Jiang,K.;Yanagida, S.J.Am.Chem.Soc.2008,130,1258.doi:10.1021/ja075704o
(4)Jung,K.S.;Lee,S.B.;Kim,Y.K.;Seo,M.H.;Hwang,W.P.; Jang,Y.W.;Park,H.W.;Jin,B.R.;Kim,M.R.;Lee,J.K. J.Photochem.Photobiol.A-Chem.2012,231,64.doi:10.1016/ j.jphotochem.2012.01.005
(5) Leijtens,T.;Ding,I.K.;Giovenzana,T.;Bloking,J.T.; McGehee,M.D.;Sellinger,A.ACS Nano 2012,6,1455.doi: 10.1021/nn204296b
(6) Caramori,S.;Cazzanti,S.;Marchini,L.;Argazzi,R.;Bignozzi, C.A.;Martineau,D.;Gros,P.C.;Beley,M.Inorg.Chim.Acta 2008,361,627.doi:10.1016/j.ica.2007.04.016
(7)Kim,J.;Koh,J.K.;Kim,B.;Ahn,S.H.;Ahn,H.;Ryu,D.Y.; Kim,J.H.;Kim,E.Adv.Funct.Mater.2011,21,4633.doi: 10.1002/adfm.v21.24
(8)Zhang,W.;Cheng,Y.M.;Yin,X.;Liu,B.Macromol.Chem. Phys.2011,212,15.doi:10.1002/macp.v212.1
(9) Chung,I.;Lee,B.;He,J.Q.;Chang,R.P.H.;Kanatzidis,M.G. Nature 2012,485,486.doi:10.1038/nature11067
(10)Natu,G.;Huang,Z.J.;Ji,Z.Q.;Wu,Y.Y.Langmuir 2012,28, 950.doi:10.1021/la203534s
(11) Prakash,T.;Ramasamy,S.Sci.Adv.Mater.2012,4,29.doi: 10.1166/sam.2012.1247
(12)Yu,M.Z.;Natu,G.;Ji,Z.Q.;Wu,Y.Y.J.Phys.Chem.Lett. 2012,3,1074.doi:10.1021/jz3003603
(13)Guo,X.Y.;Yi,P.F.;Wang,W.J.;Yang,Y.Acta Phys.-Chim. Sin.2012,28,585.[郭学益,易鹏飞,王惟嘉,杨 英.物理化学学报,2012,28,585.]doi:10.3866/PKU.WHXB201112302
(14) Benedetti,J.E.;Correa,A.A.;Carmello,M.;Almeida,L.C.P.; Goncalves,A.S.;Nogueira,A.F.J.Power Sources 2012,208, 263.doi:10.1016/j.jpowsour.2012.01.147
(15) Rachod,B.;Jutarat,S.;Nuttachai,P.;Pisorn,S.H.;Chaiyuth,S. K.;Pasit,P.Mater.Chem.Phys.2012,132,993.doi:10.1016/ j.matchemphys.2011.12.048
(16) Zhang,G.Q.;Ma,L.;Wu,Z.J.;Zhang,H.Y.;Ni,P.Acta Phys.-Chim.Sin.2009,25,555. [张国庆,马 莉,吴忠杰,张海燕,倪 佩.物理化学学报,2009,25,555.]doi:10.3866/ PKU.WHXB20090326
(17) Stephan,A.M.Eur.Polym.J.2006,42,21.doi:10.1016/ j.eurpolymj.2005.09.017
(18) Pitawala,H.M.J.C.;Dissanayake,M.A.K.L.;Seneviratne,V. A.Solid State Ionics 2007,178,885.doi:10.1016/j.ssi. 2007.04.008
(19) Lee,R.H.;Cheng,T.F.;Chang,J.W.;Ho,J.H.Colloid Polym. Sci.2011,289,817.doi:10.1007/s00396-011-2397-9
(20)Zhang,D.W.;Li,X.D.;Huang,S.M.;Wang,Z.A.;Shi,J.H.; Sun,Z.;Yin,X.J.International Journal of Nanoscience 2010, 9,257.doi:10.1142/S0219581X10006764
(21) Basak,P.;Manorama,S.V.Eur.Polym.J.2004,40,1155.doi: 10.1016/j.eurpolymj.2004.01.013
(22)Kuo,P.L.;Hou,S.S.;Lin,C.Y.;Chen,C.C.;Wen,T.C. J.Polym.Sci.Pol.Chem.2004,42,2051.doi:10.1002/pola. 20056
(23) Kumar,M.;Sekhon,S.S.Eur.Polym.J.2002,38,1297.doi: 10.1016/S0014-3057(01)00310-X
(24) Han,H.W.;Liu,W.;Zhang,J.;Zhao,X.Z.Adv.Funct.Mater. 2005,15,1940.doi:10.1002/adfm.200500159
(25)Zhou,Y.F.;Xiang,W.C.;Chen,S.;Fang,S.B.;Zhou,X.W.; Zhang,J.B.;Lin,Y.Chem.Commun.2009,3895.
(26) Liang,G.J.;Xu,J.;Xu,W.L.;Shen,X.L.;Zhang,H.;Yao,M. Polym.Compos.2011,32,511.doi:10.1002/pc.v32.4
(27) Cui,J.F.;Han,P.L.;Guo,J.H.;Yang,B.P.;Zhang,Z.L.China Synthetic Rubber Industry 2009,32,22.[崔锦峰,韩培亮,郭军红,杨保平,张志立.合成橡胶工业,2009,32,22.]
(28) Liang,G.J.;Xu,W.L.;Xu,J.;Shen,X.L.;Yao,M.Influence of Weight Ratio of CitricAcid Cross Linker on the Structure and Conductivity of the Crosslinked Polymer Electrolytes. InAdvanced Materials Research,International Symposium on Chemical Engineering and Material Properties,Shenyang, China,Nov 4-6,2011;Zhang,H.M.,Wu,B.,Eds.;Trans Tech Publications Ltd:Switzerland,2011;pp 391-392.
(29) Liang,G.J.;Xu,W.L.;Shen,X.L.;Yang,H.J.Acta Energiae Solaris Sinica 2008,29,933. [梁桂杰,徐卫林,沈小林,杨红军.太阳能学报,2008,29,933.]
(30)Yu,M.X.;Shi,Q.;Zhou,X.;Yan,Y.S.;Wan,C.R.Acta Polym. Sin.2001,5,665.[于明昕,石 桥,周 啸,严玉顺,万春荣.高分子学报,2001,5,665.]
(31) Liang,G.J.;Xu,J.;Xu,W.L.;Shen,X.L.;Bai,Z.K.;Yao,M. Polym.Eng.Sci.2011,51,2526.doi:10.1002/pen.v51.12
(32)Kang,M.S.;Kim,J.H.;Won,J.;Kang,Y.S.J.Photochem. Photobiol.A-Chem.2006,183,15.doi:10.1016/j.jphotochem. 2006.02.013
猜你喜欢
佛山陶瓷(2022年4期)2022-06-20
纺织科学研究(2021年7期)2021-08-14
山东冶金(2019年5期)2019-11-16
沈阳理工大学学报(2019年2期)2019-06-18
数字海洋与水下攻防(2018年1期)2018-10-29
现代检验医学杂志(2016年1期)2016-11-12
国外医药(抗生素分册)(2016年4期)2016-07-12
当代化工研究(2016年2期)2016-03-20
电源技术(2016年9期)2016-02-27
中国资源综合利用(2016年7期)2016-02-03
