
5-甲基胞嘧啶及胞嘧啶无辐射失活的半经典动力学模拟和CASSCF计算
2012-03-06 04:43张文英舒坤贤豆育升
物理化学学报 2012年12期
袁 帅 马 静 张文英 舒坤贤 豆育升
(重庆邮电大学生物信息研究所,重庆400065)
1 引言
脱氧核糖核酸(DNA)碱基分子由于具有π共轭结构而易吸收紫外光造成损伤,1-3然而自然界中DNA分子的光损伤产率极低,4超快失活机制是DNA碱基具有光稳定性的原因,对避免光损伤具有重要意义.单碱基激发态衰减主要通过无辐射方式进行,5-8Kohler等9使用飞秒瞬间吸收技术,在2000年第一次精确测量出DNA碱基1ππ*激发态的寿命为1 ps.荧光变频及瞬间吸收等方法对各种核苷酸Sππ*态寿命的测量10-12也得到相同的结果,即对于所有的核苷酸,碱基第一单重激发态(S1)衰减发生在亚皮秒的时间范围内.
诸多实验及理论研究认为,S1的超快衰减是由于S1与基态(S0)间的圆锥交叉(CI)所致,即CI的电子态之间超快内转换通道是核苷亚皮秒级荧光寿命的原因.9,10激发态能量对环的扭曲变形不敏感,其势能面相对较平坦;但对于基态,由于失去π键的稳定性(芳香性)后,变得非常不稳定,其能量随着环的变形程度增大而急剧增加,最终与激发态势能面相交,形成CI.量子化学计算显示核酸碱基的激发态结构是非平面的,13-23通过环扭曲引发的平面外变形达到CI.各种水平的理论研究13-23计算了所有天然碱基的CI,这些研究明确地提出,从Franck-Condon区到CI的路径几乎是无能垒的,从而使S1→S0的超快内转换得以发生.嘌呤碱基的振动弛豫模式包括沿C4=C5键的折叠产生环褶皱以及氨基的平面外扭曲运动22-24等.对于嘧啶碱基,许多研究20-25显示, C5-C6键的扭曲并伴随C5取代基的平面外运动是失活的关键.
嘧啶类碱基的激发态寿命与C5位26密切相关,这可以在C5位添加取代基以限制这一键的扭转能力来深入理解.实验26,27已经显示碱基修饰限制相关键的扭转,可以明显增长激发态寿命.例如Malone等26的研究显示,胞嘧啶(Cyt)的C5原子上的H原子被F原子取代后,S1态的寿命长达88 ps,几乎比胞嘧啶高两个数量级;5-甲基胞嘧啶的S1态寿命长达7 ps,比胞嘧啶高约10倍.26,28在尿嘧啶(Ura)和胸腺嘧啶(Thy)分子中也存在类似的现象,5-甲基的存在使得Thy的激发态衰减速率比Ura低5倍.29嘧啶C5原子上的H原子被甲基取代后,甲基的作用主要是体现在限制C5原子的扭曲;而被F原子取代后,除了限制C5原子的扭曲以外,还因其存在孤对电子,与嘧啶的芳香环形成p-π共轭,导致了更复杂的激发态性质.
正确理解一个化学反应的机理需要弄清从反应物到产物的过程中每个原子是如何运动的.然而对于碱基体系而言,受限于过于简单的反应坐标,从头算和密度泛函理论(DFT)计算往往难以顾及分子整体的运动.为了克服这一不足,本文采用半经典动力学模型模拟Cyt和5m-Cyt激发态衰减过程的每一个反应自由度以及能量随时间的变化,并且通过和完全活性空间自洽场(CASSCF)计算结果对比,从微观角度解释了5m-Cyt比Cyt的S1激发态寿命更长的原因.
2 模型与方法
本文采用的模拟激光诱导碱基光化学反应的动力学模型称为半经典电子-辐射-离子动力学(SERID)模型.所谓半经典方法是指对电子运动由含时Schrödinger方程确定,而辐射场和核运动则由经典力学处理.这种模拟方法考虑激光场对电子结构的作用,并采用非绝热动力学的近似方法和紧束缚近似来建立模型.其方法和原理已有详细报道,30,31此处不再介绍.近年来该模型成功地模拟了几个DNA碱基分子光化学反应,包括腺嘌呤(Ade)无辐射超快失活,32Thy、Cyt的光致二聚反应33,34及Thy二聚体的光致解离35反应,并描述了堆积的Ade体系、36-38Thy体系39和Ade-Thy体系40,41形成长寿命的激基复合物和成键的激基复合物的现象.这些模拟结果都与实验现象吻合,并且解释了许多实验无法说明的性质.
由于SERID模型是基于平均场理论,缺乏明确的电子态,而且紧束缚密度泛函(DFTB)方法对于小分子体系的能量计算误差较大,因此采用合理的量子化学模型对研究体系进行计算是十分必要的.例如,Bu及其合作者42,43采用含时密度泛函(TDDFT)和单激发组态相互作用(CIS)等方法很好地描述了Watson-Crick型碱基对之间的电荷转移特征.我们采用CASSCF方法计算了Cyt和5m-Cyt的势能面,并与模拟结果比较.Cyt和5m-Cyt分子结构类似,完全的活性空间包括8个π轨道和O7、N3的孤对电子,总共为14电子10轨道.由于S1态主要是ππ*激发,故我们排除两对孤对电子,并且将布居数大于1.99的轨道移出活性空间,所以最终选择的活性空间为CAS(8,7),在6-311+g(d)的基组水平计算了Cyt和5m-Cyt的垂直激发能并优化了它们的S0、S1和CI的结构.
本研究的半经典动力学模拟采用的是自行开发的SERID程序,CASSCF计算采用的是Gaussian 0944量子化学程序包,所有计算在浪潮XEON/E7(32核心/128G内存)服务器上完成.
3 结果与讨论
选择频率为4.8 eV,最大半波宽(FWHM)为25 fs的激光脉冲照射5m-Cyt和Cyt分子,脉冲频率与实验中采用的267 nm的激发光波长26,28相吻合.我们对10个不同初始构型进行模拟,得到300余条轨迹.模拟发现,超过70%的轨迹中5m-Cyt分子激发-失活过程的寿命为5-8 ps,与实验现象26,28符合,远长于Cyt的激发态寿命(模拟结果为200-800 fs).此处选择辐射通量密度为228.43 J·m-2的5m-Cyt的一条失活轨迹进行报道.
图1描述了5m-Cyt分子激发态失活的快照.脉冲作用之前分子保持平面构型(图1a);激光作用以后,5m-Cyt的芳香体系被破坏,产生一定程度的扭曲,甲基和H6原子朝不同方向振动(图1b);此后分子保持扭曲状态,甲基和H6原子朝相同和不同方向振动(图1(c,d,e,f,h));7146 fs时,甲基振动幅度最大,体系失活至基态(图1g);失活后,分子恢复平面结构(图1h).从激光停止作用到7146 fs时失活,激发态寿命约为7 ps.
图2描述了C5-C6键长随时间的变化.初始时刻C5-C6键长为0.139 nm,是典型的共轭体系C=C双键键长.激光作用以后,由于5m-Cyt分子中π被激发后π键被破坏,C5-C6转变为具有σ键性质,其键长延长至平均0.154 nm.7146 fs时由于电子衰减, 5m-Cyt分子返回基态,C5-C6恢复初始的双键结构.

图1 5m-Cyt分子激发态失活过程的快照Fig.1 Snapshots for the deactivation of the excited state of 5-methyl cytosine t/fs:(a)0,(b)1955,(c)3088,(d)3950,(e)5073,(f)6034,(g)7146,(h)7480

图2 5m-Cyt分子C5-C6键长随时间的变化图Fig.2 Variations with time of C5-C6bond lengths of 5m-Cyt molecule

图3 5m-Cyt分子θ(N3-C4-C5-C6)二面角和能隙(ΔELUMO-HOMO)随时间的变化Fig.3 Variations with time of dihedral angle θ(N3-C4-C5-C6)and energy gap(ΔELUMO-HOMO)of 5m-Cyt molecule
图3描述了二面角θ(N3-C4-C5-C6)随时间的变化.该二面角的变化表征了C5、C6原子的扭曲变形程度.从图3看出,激光作用之前,5m-Cyt分子保持平面构型,θ(N3-C4-C5-C6)=0°;激光作用以后, θ(N3-C4-C5-C6)二面角发生显著改变,表明C5-C6键发生扭曲.θ(N3-C4-C5-C6)的值波动范围约为±40°,意味着C5-C6键发生强烈的振动.ΔELUMO-HOMO是最低未占据分子轨道(LUMO)与最高占据分子轨道(HOMO)的能隙,代表轨道非绝热耦合的程度. ΔELUMO-HOMO初始值为4.2 eV,随着C5-C6键开始扭曲而减小,意味着C5-C6键扭曲有利于体系发生非绝热衰减.但是从图3看出,当甲基和H6原子向同一方向振动时,ΔELUMO-HOMO有增大的趋势(如图3(c、e)所示),表明该结构相对稳定;当甲基和H6原子向不同方向振动时,ΔELUMO-HOMO趋于减小(如图3(b、d、f、g)所示),表明该结构不稳定,有失活的可能性.在避免交叉点ΔELUMO-HOMO=0.09 eV,θ(N3-C4-C5-C6)达到42°.此时θ(N3-C4-C5-C6)角度值与模拟过程中b、f等结构相比并无显著变化,表明C5-C6键扭曲虽然与ΔELUMO-HOMO密切相关,但并非促成非绝热衰减的唯一因素.
图4中θ(N1-C6-H6-C5)和θ(C3-C4-C5-C7)二面角分别表征5m-Cyt分子的H6原子和甲基的平面外扭曲程度.θ(N1-C6-H6-C5)自激光作用以后,迅速从180°降为120°,并一直保持在此值上下波动;7146 fs时达到最小值90°,表明此时H6原子几乎和环面垂直.θ(C3-C4-C5-C7)则在模拟过程中一直在130°-240°之间波动,表明甲基在模拟过程中一直在上下振动;衰减时刻θ(C3-C4-C5-C7)达到其最大值254°(意味着甲基与环面近似垂直),明显大于模拟过程中θ(C3-C4-C5-C7)的振动范围.这意味着甲基和H6原子的环面外振动对5m-Cyt分子的失活起着决定性的作用,是到达超快无辐射失活的CI点的必需途径.
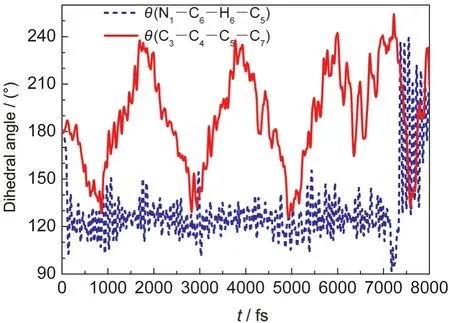
图4 5m-Cyt分子的θ(N1-C6-H6-C5)和θ(C3-C4-C5-C7)二面角与时间关系Fig.4 Variation with time of the θ(N1-C6-H6-C5)and θ(C3-C4-C5-C7)dihedral angles of 5m-Cyt molecule

图5 Cyt分子θ(H6-C6-C5-H5)和θ(N3-C4-C5-C6)二面角随时间的变化Fig.5 Variations of θ(H6-C6-C5-H5)and θ(N3-C4-C5-C6)dihedral angles of cytosine molecule with time
由于Cyt分子的模拟结果与5m-Cyt基本相似,主要差别在激发态寿命不同,此处仅简单介绍.图5描述了Cyt分子的θ(H6-C6-C5-H5)和θ(N3-C4- C5-C6)二面角随时间的变化,这两个二面角分别表征H5、H6原子平面外振动的幅度和C5-C6键的扭曲程度.该模拟轨迹的脉冲频率为4.8 eV,辐射通量密度为214.52 J·m-2.该轨迹中Cyt分子在210 fs时发生非绝热跃迁失活至基态.在避免交叉点时,θ(H6-C6-C5-H5)达到了其最小值-152°,而θ(N3-C4-C5-C6)=-39°,与其它时刻相比变化不明显.因此, Cyt的ΔELUMO-HOMO对C5-C6键的扭曲不敏感,促使激发态衰减的振动主要是H5、H6原子平面外振动.由于C5、C6原子强烈的扭曲,其pz轨道与π体系不再耦合,并保持单占据,从而具有双自由基的性质. Zgierski等45的理论计算提出,Cyt分子的ππ*态将沿着C5-C6扭曲的坐标演化为“双自由基态”而失活.“双自由基态”中,H5、H6原子几乎与环面垂直,并且指向相反方向.从结构来看,本文模拟的结果与量子化学计算符合得很好.
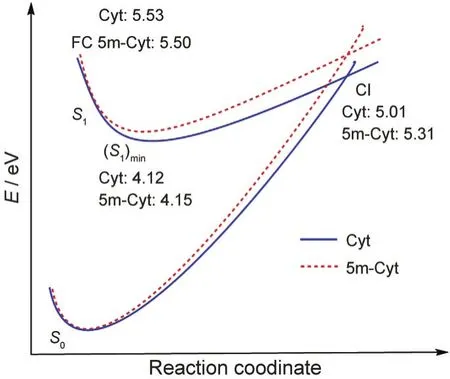
图6 Cyt和5m-Cyt分子的S0-S1势能面Fig.6 S0-S1potential energy surfaces of Cyt and 5m-Cyt molecules
图6描述了CASSCF(8,7)/6-311+g(d)水平计算的Cyt和5m-Cyt分子的S0和S1势能面.Cyt分子的垂直激发能为5.53 eV,比实验值4.6 eV46高,原因在于未包含动态相关能,但是与Robb等22在CASSCF (14,10)水平计算得到的5.21 eV接近.虽然本文的CASSCF计算比较粗糙,但是定性比较能垒高低是足够的.Cyt垂直激发能和绝热激发能和5m-Cyt分子都接近,但是CI的能量比后者低0.3 eV,意味着5m-Cyt分子达到CI比Cyt慢,也验证了模拟结果.
4 结论
采用半经典动力学方法对激光诱导下5m-Cyt和Cyt分子的最低激发态的失活过程进行了模拟.模拟脉冲频率与实验值相当,为4.8 eV,FWHM=25 fs.激光作用以后,5m-Cyt和Cyt分子的C5-C6键发生扭曲,在甲基(或H5)和H6原子平面外振动幅度最大形成“双自由基态”,体系发生非绝热失活.失活时甲基(或H5)和H6原子几乎与环面垂直.由于甲基的空间位阻效应,5m-Cyt形成“双自由基态”的时间约为7 ps,远长于Cyt的210 fs.CASSCF计算证实, 5m-Cyt的(S1/S0)CI能量比Cyt高0.3 eV,意味着5m-Cyt的失活需要克服更高的能垒,因此其S1激发态寿命更长.
(1)Beukers,R.;Eker,A.P.M.;Lohman,P.H.M.DNA Repair 2008,7,530.doi:10.1016/j.dnarep.2007.11.010
(2)Melnikova,V.O.;Ananthaswamy,H.N.Mutat.Res.2005,571, 91.doi:10.1016/j.mrfmmm.2004.11.015
(3) Cadet,J.;Sage,E.;Douki,T.Mutat.Res.2005,571,3.doi: 10.1016/j.mrfmmm.2004.09.012
(4) Mouret,S.;Baudouin,C.;Charveron,M.;Favier,A.;Cadet,J.; Douki,T.Proc.Natl.Acad.Sci.U.S.A.2006,103,13765.doi: 10.1073/pnas.0604213103
(5) Crespo-Hernández,C.;Cohen,B.;Hare,P.;Kohler,B.Chem. Rev.2004,104,1977.
(6) Shukla,M.;Leszczynski,J.J.Biomol.Struct.Dyn.2007,25, 93.doi:10.1080/07391102.2007.10507159
(7) Saigusa,H.J.Photochem.Photobiol.C 2006,7,197.doi: 10.1016/j.jphotochemrev.2006.12.003
(8) de Vries M.;Hobza,P.Annu.Rev.Phys.Chem.2007,58,585. doi:10.1146/annurev.physchem.57.032905.104722
(9) Pecourt,J.M.L.;Peon,J.;Kohler,B.J.Am.Chem.Soc.2000, 122,9348.doi:10.1021/ja0021520
(10) Crespo-Hernandez,C.E.;Cohen,B.;Hare P.M.;Kohler,B. Chem.Rev.2004,104,1977.doi:10.1021/cr0206770
(11) Peon,J.;Zewail,A.H.Chem.Phys.Lett.2001,348,255.
(12) Gustavsson,T.;Sharonov,A.;Markovitsi,D.Chem.Phys.Lett. 2002,351,195.doi:10.1016/S0009-2614(01)01375-6
(13) Shukla,M.K.;Leszczynski,J.J.Biomol.Struct.Dyn.2007,25, 93.doi:10.1080/07391102.2007.10507159
(14) Conti,I.;Altoè,P.;Stenta,M.;Garavelli,M.;Orlandi,G.Phys. Chem.Chem.Phys.2010,12,5016.
(15) Serrano-Andres,L.;Merchan,M.J.Photochem.Photobiol.C 2009,10,21.doi:10.1016/j.jphotochemrev.2008.12.001
(16) Langer,H.;Doltsinis,N.L.J.Chem.Phys.2003,118,5400. doi:10.1063/1.1555121
(17) Canuel,C.;Mons,M.;Piuzzi,F.;Tardivel,B.;Dimicoli,I.; Elhanine,M.J.Chem.Phys.2005,122,074316.doi:10.1063/ 1.1850469
(18) Zechmann,G.;Barbatti,M.J.Phys.Chem.A 2008,112,8273. doi:10.1021/jp804309x
(19) Karunakaran,V.;Kleinermanns,K.;Improta,R.;Kovalenko,S. A.J.Am.Chem.Soc.2009,131,5839.doi:10.1021/ja810092k
(20)Kwok,W.M.;Ma,C.;Phillips,D.L.J.Am.Chem.Soc.2008, 130,5131.doi:10.1021/ja077831q
(21) Zechmann,G.;Barbatti,M.J.Phys.Chem.A 2008,112,8273. doi:10.1021/jp804309x
(22) Ismail,N.;Blancafort,L.;Olivucci,M.;Kohler,B.;Robb,M.A. J.Am.Chem.Soc.2002,124,6818.doi:10.1021/ja0258273
(23) Merchán,M.;González-Luque,R.;Climent,T.;Serrano-Andrés,L.;Rodríguez,E.;Reguero,M.;Peláez,D.J.Phys. Chem.B 2006,110,26471.doi:10.1021/jp066874a
(24) Zgierski,M.Z.;Patchkovskii,S.;Fujiwara,T.;Lim,E.C. Chem.Phys.Lett.2007,440,145.doi:10.1016/j.cplett. 2007.04.017
(25) Serrano-Andrés,L.;Merchán,M.;Borin,A.C.Chem.Eur.J. 2006,12,6559.
(26) Malone,R.J.;Miller,A.M.;Kohler,B.Photochem.Photobiol. 2003,77,158.doi:10.1562/0031-8655(2003)077<0158: SESLOC>2.0.CO;2
(27) Zgierski,M.Z.;Patchkovskii,S.;Fujiwara,T.;Lim,E.C. Chem.Phys.Lett.2007,440,145.doi:10.1016/j.cplett. 2007.04.017
(28) Sharonov,A.;Gustavsson,T.;Marguet,S.;Markovitsi,D. Photochem.Photobiol.Sci.2003,2,1.doi:10.1039/b211055e
(29) Gustavsson,T.;Banyasz,A.;Lazzarotto,E.;Markovitsi,D.; Scalmani,G.;Frisch,M.J.;Barone,V.;Improta,R.J.Am. Chem.Soc.2006,128,607.doi:10.1021/ja056181s
(30) Dou,Y.S.;Torralva,B.R.;Allen,R.E.Chem.Phys.Lett.2004, 392,352.doi:10.1016/j.cplett.2004.05.087
(31) Dou,Y.S.;Torralva,B.R.;Allen,R.E.J.Mod.Optics.2003, 50,2615.
(32)Lei,Y.;Yuan,S.;Dou,Y.;Wang,Y.;Wen,Z.J.Phys.Chem.A 2008,112,8497.doi:10.1021/jp802483b
(33) Zhang,W.;Yuan,S.;Li,A.;Dou,Y.;Zhao,J.;Fang,W.J.Phys. Chem.C 2010,114,5594.doi:10.1021/jp907290f
(34)Yuan,S.;Zhang,W.Y.;Liu,L.H.;Dou,Y.;Fang,W.H.;Lo,G. V.J.Phys.Chem.A 2011,115,13291.
(35)Dou,Y.;Xiong,S.;Wu,W.F.;Yuan,S.;Tang,H.J.Photochem. Photobiol.B 2010,101,31.doi:10.1016/j.jphotobiol. 2010.06.008
(36)Zhang,W.Y.;Yuan,S.;Wang,Z.;Qi,Z.;Zhao,J.;Dou,Y.;Lo, G.Chem.Phys.Lett.2011,506,303.doi:10.1016/j.cplett. 2011.03.024
(37)Yuan,S.;Zhang,W.Y.;Li,A.Y.;Zhu,Y.M.;Dou,Y.S.Acta Phys.-Chim.Sin.2011,27,824. [袁 帅,张文英,李安阳,朱义敏,豆育升.物理化学学报,2011,27,824.]doi:10.3866/ PKU.WHXB20110341
(38)Dou,Y.S.;Zhao,W.H.;Yuan,S.;Zhang,W.Y.;Tang,H.Sci. Chin.Chem.2012,55,1377.[豆育升,赵文辉,袁 帅,张文英,唐 红.中国科学:化学,2012,55,1377.]doi:10.1007/ s11426-012-4578-x
(39)Dou,Y.S.;Li,W.;Yuan,S.;Zhang,W.Y.;Li,A.Y.;Shu,K.X.; Tang,H.Acta Phys.-Chim.Sin.2011,27,2559.[豆育升,李 伟,袁 帅,张文英,李安阳,舒坤贤,唐 红,物理化学学报,2011,27,2559.]doi:10.3866/PKU.WHXB20111115
(40)Zhang,W.Y.;Ma,J.;Yuan,S.;Shu,K.X.;Dou,Y.S.Acta Phys.-Chim.Sin.2012,28,1676.[张文英,马 静,袁 帅,舒坤贤,豆育升,物理化学学报,2012,28,1676.] doi:10.3866/PKU.WHXB201205041
(41)Dou,Y.S.;Yuan,S.;Zhang,W.Y.;Tang,H.;Lo,G.V.Mol. Phys.2012,110,1517.doi:10.1080/00268976.2012.663944
(42) Zhang,L.B.;Bu,Y.X.J.Phys.Chem.B 2008,112,10723.doi: 10.1021/jp802556a
(43) Zhang,L.B.;Li,H.F.;Li,J.L.;Chen,X.H.;Bu,Y.X. J.Comput.Chem.2009,31,825.
(44) Frisch,M.J.;Trucks,G.W.;Schlegel,H.B.;et al.Gaussian 09, Revision C.01;Gaussian Inc.:Wallingford,CT,2010.
(45) Zgierskia,M.Z.;Patchkovskii,S.J.Chem.Phys.2005,123, 081101.doi:10.1063/1.2031207
(46) Fülscher,M.P.;Roos,B.O.J.Am.Chem.Soc.1995,117,2089.
猜你喜欢
数学物理学报(2022年3期)2022-05-25
科学导报(2022年28期)2022-05-24
数学物理学报(2022年1期)2022-03-16
数学物理学报(2021年5期)2021-11-19
数学物理学报(2021年3期)2021-07-19
汕头大学学报(自然科学版)(2020年4期)2020-12-14
发明与创新·大科技(2019年6期)2019-09-06
原子与分子物理学报(2015年3期)2015-11-24
读写算·教研版(2014年12期)2014-09-01
原子与分子物理学报(2014年1期)2014-03-20
