
TMnCOTn+1层状团簇的结构及其电磁性质
2012-01-29 10:32张秀云刘拥军
扬州大学学报(自然科学版) 2012年1期
韩 玖,张秀云,刘拥军
(扬州大学 物理科学与技术学院,江苏 扬州 225002)
金属原子和有机分子环构成的化合物在光电子器件、催化剂、生物传感器等方面展现出诱人的应用前景,其中有机金属层状化合物由于其独特的结构和电磁性质而被广泛关注.[1-8]例如,由苯环和V原子组成的VnBzn+1团簇及其一维无限长的纳米线已有较多的报道.[8-11]事实上,V原子可以与不同的有机分子形成稳定的层状团簇,包括VnBzm,其中1975年已合成出V原子与C5H5形成的VCp2[8],它的电子结构被预测为4A1′[10];但是多层的 VnCpm到目前为止仍然没有合成出[9].类似地,还有一维无限长的[VBz]∞和 [VCp]∞已经被预测,是一种半金属的铁磁体,并且它们有限的团簇在Li电极上具有很高的自旋极化率.[7]13958尽管由过渡金属原子组成的团簇已引起人们很大的兴趣,但是近年来报道的由4fLn-COT组成的团簇也表现出重要的性质.例如,Stern-Gerlach的磁性偏转实验[11-12]已经说明 LnnCOTm(Ln:Eu,Tb,Ho,Tm;n=1~7)表现出大的磁矩.特别是EunCOTn+1团簇,其磁矩随着团簇尺寸的增加呈现线性增加,并且18层的EunCOTn+1纳米线已经通过激光蒸气的方法合成出.[13]事实上,大量的光谱表明LnnCOTn+1(Ln:Ce,Nd,Eu,Ho,Yb)团簇是稳定的.[14]然而,这些研究仅局限于镧系金属原子,尽管由过渡金属阳离子与1,3,5,7-COT形成的化合物 M+(C8H8)1,2[15](M:V,Fe,Ni,Ag)已经合成出,但对由过渡金属原子与COT组成的团簇却研究很少.为此,笔者将应用密度泛函理论的方法来探究TMnCOTn+1(TM:Ti,Zr,Hf)的几何结构和电磁性质.
1 计算方法
本文所有的计算均通过Dmol3软件[16]进行,交换关联函数采取广义梯度近似(GGA)中的PW91[17],选择的基组是双数值极化基(DNP),对核外电子的处理采用态密度函数离子实赝势(DSPP),以考虑相对论效应.对于自洽场计算,总能和电子密度的收敛标准是2.72×10-5eV,结构优化收敛标准中力的变化小于0.544eV·nm-1,位置移动变化小于0.000 5nm,总能变化小于2.72×10-4eV,梯度的收敛标准是2.72×10-2eV.在优化过程中,所有结构都未采取对称性限制.
2 计算结果和讨论
2.1 TMnCOTn+1团簇的结构
图1列出了TMCOT和TMnCOTn+1的几何结构和TM为Ti时COT环上的Mulliken电荷.对于TMCOT团簇,通过在其COT环上添加一个TM原子就形成了稳定的半层状团簇,当TM为Ti时,其几何对称性为C2v,自旋多重态是三重态,其中Ti原子距离COT中心0.138 1nm,C—C和C—H键的长度分别为0.140 9,0.107 6nm;而当TM分别为Zr,Hf时,自旋态都是单重态,Zr,Hf原子距离COT的中心分别为0.161 1,0.158 5nm,C—C键长分别为0.148 0,0.141 9nm,C—H键长分别为0.109 7,0.109 8nm.
对于TMCOT2团簇,当TM为Ti时,其几何对称性为D4h,自旋态是单重态,其中Ti原子距离COT中心0.167 2nm,C—C和C—H键的长度分别为0.138 0,0.107 4nm.Ti原子位于COT环的中心,该结果与EuCOT2的结论[18]相一致,但是与FeCOT2[19]不一致,这是因为Fe原子并不是位于两个相邻的COT环的中心;当TM为Zr时,自旋态是单重态,Zr原子距离COT的中心分别是0.178 0,0.179 2nm,说明Zr原子并不是位于两个COT环的几何中心;当TM为Hf时,自旋态是单重态,Zr原子距离COT的中心0.177 8nm,C—C和C—H 键的长度分别为0.141 4,0.109 0nm.
对于TM2COT3和TM3COT4,自旋态都是单重态,C—H键在COT中的距离几乎一样,约为0.107nm,C—C键之间的范围为0.139 5~0.142 7nm.对于Ti3COT4,相邻两个COT环之间的垂直距离是0.342 0,0.338 4nm,而Ti—C之间的平均距离是0.25nm,说明Ti原子并不是确切地位于两个COT环的中间.例如,对于Ti2COT3,Ti原子与终端的COT环之间的距离0.153 8nm明显地比Ti原子与中心的COT环的距离0.191 8nm短.这个特性说明Ti原子更加倾向于终端的COT环在Ti2COT3和Ti3COT4中.这是由于边缘的COT环内外非对称分布的电荷密度(图1)造成的,从而导致 Ti原子轻微地偏向于边缘的COT环,该结论与FenCOTn+1(n=2~4)[19]11948和EunCOTn+1(n=2~4)[18]2517是一致的.总之,上述分析表明 TM2COT3和 TM3COT4中的 TM 原子并不位于两个COT环的中心,并且对于ZrCOT2,Zr原子也不位于两个COT环的中心.
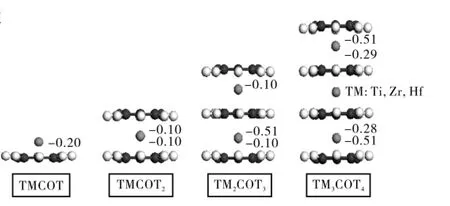
图1 优化的TMCOT,TMnCOTn+1(n=1~3)的结构和TM为Ti原子时COT环上的Mulliken电荷Fig.1 Optimized structures of TMCOT,TMnCOTn+1(n=1~3)and the Mulliken charge of COT for Ti atom
2.2 TMnCOTn+1的 HOMO-LUMO能隙
HOMO-LUMO能隙反映团簇电子结构的稳定性.图2(a)给出了 HOMOLUMO能隙随团簇尺寸的变化趋势.有趣的是,这些团簇的HOMO-LUMO能隙呈现奇偶的变化趋势,例如TMCOT和TM2COT3表现出较大的能隙,TMCOT2和TM3COT4表现出较小的能隙.这些说明TMCOT和TM2COT3具有相对较稳定的电子结构.但是当TM为Ti原子时,这些化合物的HOMO-LUMO能隙比较小;当TM为Zr,Hf时,能隙相对于Ti原子比较大,说明Zr,Hf原子的团簇电子结构比较稳定.总体而言,无论是金属原子Ti的化合物,还是Zr,Hf的化合物,TM2COT3团簇的HOMO-LUMO能隙总是最大的,分别为1.358,1.28,1.46eV,这似乎说明TM2COT3相对于其他化合物的电子结构将更加稳定.
2.3 TinCOTn+1团簇的稳定性
TMnCOTn+1团簇对应单个TM原子和COT分子的结合能的计算方法如下:
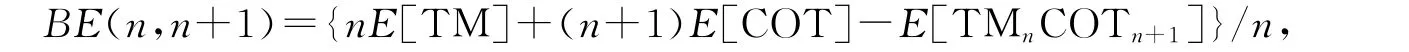
E[TMnCOTn+1],E[COT],E[TM]分别表示 TMnCOTn+1团簇、COT 环、TM 原子的总能量.图2(b)给出计算得到的各个尺寸团簇的结合能,其中TiCOT的结合能是4.284eV,而TiCOT2的结合能迅速增加到6.998eV,但是随着尺寸的进一步增大,结合能趋向于常数.这说明在大尺寸情况下,Ti原子与COT之间的结合能几乎不受团簇尺寸的影响.此外,TiCOT以及TinCOTn+1(n=1~3)的结合能从数值上看都很大,说明它们是稳定存在的;当TM为Zr,Hf时,TMCOT的结合能分别为5.61,5.78eV,而TMCOT2的结合能迅速增大到9.85,10.40eV;但是随着尺寸的进一步增大,结合能缓慢地减小.这说明在大尺寸情况下,TM原子与COT之间的结合变得相对不稳定.不论TM为何种原子,对于TMCOT2,TM2COT3,TM3COT4而言,结合能具有最大值,说明这些团簇相对于TMCOT结构更加稳定.这个结论很容易理解,这是因为TM原子在TMCOT中只连接一个COT环,并且对于大尺寸的团簇,TM原子更加倾向于边缘的COT环,所以相对于TMCOT结构更加稳定.
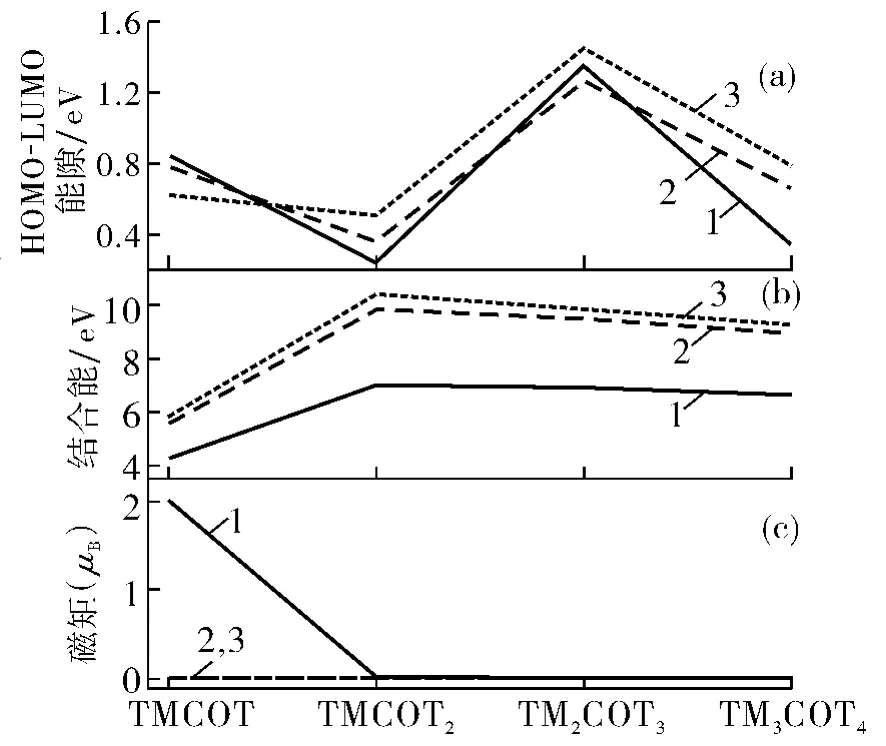
图2 TMnCOTn+1的 HOMO-LUMO能隙(a),结合能(b)和基态总磁矩(c)Fig.2 HOMO-LUMO gap(a),binding energy(b)and total MMs(c)of TMnCOTn+1
2.4 TinCOTn+1团簇的磁性质
图2(c)列出了 TMCOT和 TMnCOTn+1的总磁矩.TiCOT的总磁矩是2μB,但是增加一个COT成为TiCOT2后,团簇的总磁矩减小为0,并且随着尺寸的进一步增大,总磁矩为常数0;当TM为Zr,Hf原子时,所有团簇的磁矩均为0.为了更深刻地理解TMnCOTn+1的磁性质,图 3给出 了 TiCOT 和Ti2COT3分子的前线分子轨道图.对于TiCOT,只有很少的密度在COT环上,TiCOT的磁矩主要由Ti原子决定,从HOMO到 HOMO-5,自旋向上和自旋向下的态是非对称的,出现了两个未配对的电子,因此TiCOT团簇的总磁矩是2μB.对于TiCOT2,从HOMO到 HOMO-7,密度不仅来自COT环,而且也来自金属原子,但是自旋向上和自旋向下的状态是对称的,没有未配对的电子,所以磁矩为0.同样,对于Ti2COT3和 Ti3COT4,从 HOMO 到 HOMO-7,密度不仅来自COT环,而且还来自金属原子,但是自旋向上和自旋向下是对称的,因此总磁矩是0.
为了探究这些团簇的自旋稳定性,表1给出了计算的TiCOT和TinCOTn+1的基态与第一激发态的能量差,可以看出它们之间的能量差很小,说明这些团簇有高的自旋稳定性.例如,对于TiCOT2团簇,第一激发态仅仅比基态的能量高0.025eV,这说明TiCOT2自旋稳定性好.
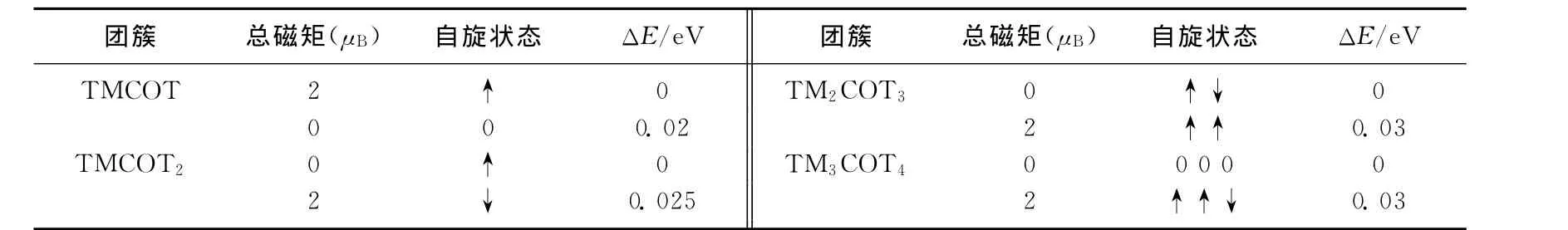
表1 TiCOT和TinCOTn+1的总磁矩、Ti原子的自旋状态、基态与激发态的能量差Tab.1 Total MMs,Ti atom’s spin state,energy difference between ground and excited state of TiCOT and TinCOTn+1
[1]HOSHINO K,KURIKAWA T,TAKEDA H,et al.Structures and ionization energies of sandwich clusters(Vn(benzene)m)[J].J Phys Chem,1995,99(10):3053-3055.
[2]WEIS P,KEMPER P R,BOWERS M T.Structures and energetics of Vn(C6H6)m+clusters:evidence for a quintuple-decker sandwich[J].J Phys Chem A,1997,101(44):8207-8213.
[3]WANG Jin-lan,ACIOLI P H,JELLINEK J.Structure and magnetism of VnBzn+1sandwich clusters[J].J Am Chem Soc,2005,127(9):2812-2813.
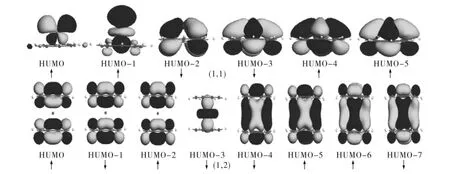
图3 TiCOT和TiCOT2团簇的前线分子轨道Fig.3 The front molecular orbital of TiCOT and TiCOT2
[4]MIYAJIMA K,YABUSHITA S,KNICKELBEIN M B,et al.Stern-gerlach experiments of one-dimensional metal-benzene sandwich clusters:Mn(C6H6)m(M=Al,Sc,Ti,and V)[J].J Am Chem Soc,2007,129(27):8473-8480.
[5]XIANG Hong-jun,YANG Jin-long,HOU J G,et al.One-dimensional transition metal-benzene sandwich polymers:possible ideal conductors for spin transport[J].J Am Chem Soc,2006,128(7):2310-2314.
[6]MASLYUK V V,BAGRETS A,MEDED V,et al.Organometallic benzene-vanadium wire:a one-dimensional half-metallic ferromagnet[J].Phys Rev Lett,2006,97(9):097201:1-4.
[7]SHEN Lei,YANG Shuo-wang,NG M F,et al.Charge-transfer-based mechanism for half-metallicity and ferro-magnetism in one-dimensional organometallic sandwich molecular wires[J].J Am Chem Soc,2008,130(42):13956-13960.
[8]GARD E,HAALAND A,NPVAK D P,et al.The molecular structures of dicyclopentadienylvanadium,(C5H5)2V,and dicyclopentadienylchromium,(C5H5)2Cr,determined by gas phase electron diffraction[J].J Organomet Chem,1975,88(2):181-189.
[9]NAGAO S,NAKAJIMA A,KAYA K,et al.Multiple-decker sandwich poly-ferrocene clusters[J].J Am Chem Soc,2000,122(17):4221-4222.
[10]XU Zhen-feng,XIE Yao-ming,FENGwen-lin,et al.Systematic investigation of electronic and molecular structures for the first transition metal series metallocenes M(C5H5)2(M=V,Cr,Mn,Fe,Co,and Ni)[J].J Phys Chem A,2003,107(15):2716-2729.
[11]MIYAJIMA K,KNICKELBEIN M B,NAKAJIMA A.Magnetic properties of lanthanide organometallic sandwich complexes produced in a molecular beam [J].Polyhedron,2005,24(16/17):2341-2345.
[12]MIYAJIMA K,KNICKELBEIN M B,NAKAJIMA A.Stern-gerlach study of multidecker lanthanide-cyclooctatetraene sandwich clusters[J].J Phys Chem A,2008,112(3):366-375.
[13]TAKEGAMI R,HOSOYA N,SUZUMURA J,et al.Geometric and electronic structures of multiple-decker one-end open sandwich clusters:Eun(C8H8)n-(n=1~4)[J].J Phys Chem A,2005,109(11):2476-2486.
[14]KURIKAWA T,NEGISHI Y,HAYAKAWA F,et al.Multiple-decker sandwich complexes of lanthanide-1,3,5,7-cyclooctatetraene[Lnn(C8H8)m](Ln=Ce,Nd,Eu,Ho,and Yb);localized ionic bonding structure[J].J Am Chem Soc,1998,120(45):11766-11772.
[15]JAEGER T D,DUNCAN M A.Photodissociation processes in transition-metal cation complexes with cyclooctatetraene[J].J Phys Chem A,2004,108(51):11296-11301.
[16]DELLEY B.An all-electron numerical method for solving the local density functional for polyatomic molecules[J].J Chem Phys,1990,92(1):508-517.
[17]PERDEW J P,WANG Yue.Accurate and simple analytic representation of the electron-gas correlation energ[J].Phys Rev B,1992,45(23):13244-13249.
[18]ZHANG Xiu-yun,NG M F,WANG Yan-biao,et al.Theoretical studies on structural,magnetic,and spintronic characteristics of sandwiched EunCOTn+1(n=1~4)clusters[J].ACS Nano,2009,3(9):2515-2522.
[19]HUANG Jing,LI Qun-xiang,XU Ke,et al.Electronic,magnetic,and transport properties of Fe-COT clusters:a theoretical study[J].J Phys Chem C,2010,114(27):11946-11950.
猜你喜欢
China’s foreign Trade(2021年6期)2021-12-26
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
中学化学(2017年6期)2017-10-16
中学化学(2017年6期)2017-10-16
汽车与新动力(2017年3期)2017-06-29
中学化学(2017年2期)2017-04-01
试题与研究·高考理综化学(2016年3期)2017-03-28
中华奇石(2015年7期)2015-07-09
