
含能材料的量子化学计算与分子动力学模拟综述
2012-01-28 03:03居学海叶财超徐司雨
火炸药学报 2012年2期
居学海,叶财超,徐司雨
(1.南京理工大学化工学院,江苏 南京210094;2.西安近代化学研究所燃烧与爆炸技术重点实验室,陕西 西安710065)
引 言
近30年来,量子化学计算方法和分子模拟技术、以及计算机技术的飞速发展,对材料科学的发展产生了深刻影响。材料科学与物理、化学、数学和工程力学等学科相互交叉与渗透,产生了计算材料学这一新兴学科。通过对材料组分和材料结构的模型化计算实现对材料设计、制备、加工、结构和性能等参数或过程的定量描述,建立材料结构与性能之间的关系,并最终按指定目标设计新材料。研究对象的空间和时间尺度不同,计算材料学的研究范围、深度和方法也随之不同。按材料的空间尺度大体可划分为4个层次:电子/原子层次、微观、介观和连续体(宏观)层次;或简单地分为微观、介观和宏观3个层次。计算方法依次为量子力学方法(半经验分子轨道法、第一性原理)、第一性原理分子动力学模拟(QMD)、经典分子动力学(MD)和蒙特卡罗模拟、耗散粒子动力学模拟、相场动力学和原胞自动化方法、有限元和有限差分法等。一般来说,通过电子及原子层次计算实现对材料本质的认识,是材料设计的科学基础。其研究方法主要为量子力学(在化学和材料学领域,常称量子化学)和分子动力学模拟。量子化学(QC)的研究对象为平衡态、单分子或几个分子组成的体系、周期性体系(晶胞或超晶胞);不适用于动力学过程和有温度压力变化的体系。分子动力学模拟描述原子核的运动过程,求得系统的结构和性质,模拟结果既包含系统的静态性质,又包含动态特性;经典(经验势)分子动力学可研究上百万甚至千万个原子的体系,但不适用于有电子转移、原子变价的过程。后者须采用第一性原理分子动力学。本文主要介绍量子化学和经典分子动力学模拟在含能材料研究中的应用及进展。
1 热力学性质及爆轰性能
含能材料的爆轰性能与爆热密切相关,而爆轰过程的反应热可由爆轰反应各物质的生成热(HOFs)直接求得。通过选用适当的方法,准确计算含能材料分子的生成热是量子化学的一大优势。最早使用半经验分子轨道方法可迅速并直接计算出生成热。但这些半经验方法主要依据一些代表性小分子和烃类的热力学和光谱数据进行参数化,对含多种取代基或特殊结构的含能材料生成热的计算往往带来较大的误差。一些改进的半经验方法,如PDDG(Pairwise Distance Directed Gaussian modification)改进法,可减少误差。比较了622种含CHNO化合物生成热计算值与实验值的绝对误差,PM3方法为18.4kJ/mol,PDDG/PM3方法为13.4kJ/mol[1]。
随着计算技术的发展,第一性原理计算(从头计算和密度泛函)逐渐取代半经验分子轨道法在含能材料中普遍运用。但第一性原理只能求得分子的总能量,无法直接计算生成热。这就要求设计等键反应,利用参考物的实验生成热,借助Hess定律,求得目标分子的生成热。在设计等键反应时,根据键分离规则把分子分解成一系列与所求物质具有相同化学键类型的小分子(已知生成热)。为减少误差,设计等键反应时应尽量使母体骨架或原有化学键保持不变。
第一性原理计算方法很多。含能材料闭壳层限制性Hartree-Fock(HF)计算可得到准确的分子几何构型,但求得的能量与实验误差通常在200kJ/mol以内,个别误差高达700kJ/mol[2]。因HF 方法忽略电子相关效应,导致能量值出现系统正误差。为此,可用微扰法(MP2和MP4等)、多组态法(如CISD、CCSD 和CASSCF)及密度泛函理论(DFT)法(如B3LYP、B3PW91)校正电子相关效应。微扰法和DFT 法计算的能量误差通常在40kJ/mol以内,而多组态法计算的误差一般小于5~8kJ/mol[2]。多组态法即使对中等体系的计算量也非常大,因此通常用DFT 法处理电子相关问题。虽然DFT 法的能量绝对误差40kJ/mol是一个不小的值,但如前所述,生成热的计算通过设计等键反应来实现,反应物与产物的能量误差大部分相互抵消。DFT 泛函的选用,通常要以同类型化合物的准确实验生成热为基准,比较并检验各种泛函对该类化学物的计算结果,以确定合适方案。
量子化学计算出的生成热是气相生成热,结合遗传算法(GA)、静电势法和神经网络法(NN)等求得固体的升华热后,方可求得固相生成热。静电势法的计算误差小,在含能材料升华热计算中应用较广[3]。对有机分子组成的固体,其升华热取决于分子间相互作用能,相互作用能越大则升华热越大。因此,升华热与分子表面的静电势有关。值得一提的是,新近基于1 384个化合物(含172个化学基团)升华热所建立的3层前馈神经网络计算模型[4],其升华热的计算值与实验结果的相关系数平方、平均误差和均方根误差分别为0.985 4,3.54%和4.21kJ/mol。远小于静电势法的误差11.7kJ/mol,具有一定的适用性和精确性。对于含能材料,该方法能否准确预测化合物的升华热还有待检验。
用Kamlet经验公式[5]预测爆速和爆压时,不仅需要生成热值,还要有密度值。基于化合物优化构型,用Monte-Carlo 法求得分子周边0.001e/bohr3等电子密度面所包围的体积,得到摩尔体积(V)。因该法求得的体积值在较大范围内波动,通常要取100次以上平均值。由相对分子质量与平均摩尔体积之比求得理论密度(ρ)。计算表明,在B3LYP/6-31G**水平下计算的理论密度与实测值吻合较好[6]。表1列出一些含能材料分子的气相生成热、爆速和爆压。结果表明,计算值与实验值相吻合。用第一性原理计算结合等键反应不仅求得生成热,还能求得基团相互作用能和环张力[7-8],从分子水平阐明结构与性能的关系。

表1 一些含能材料的气相生成热(ΔfHgas)、爆压和爆速Table 1 Heats of formation,detonation pressure and velocity for some energetic materials
2 分解反应机理
含能材料的爆燃过程是以凝聚相和气相化学反应(含气固界面反应)为基础的多组分多阶段过程。量子化学计算是通过研究热分解过程各基元反应历程,建立反应物、过渡态(包括中间体)和产物之间的联系。近年来,第一性原理方法广泛应用于研究爆燃条件下的热分解基元反应。例如,量子化学计算揭示氨基四唑的分解途径主要有两种方式[10]:两键断裂开环分解成RN3和NH2CN 及一键断裂开环生成叠氮化物,该叠氮化物再失去一分子N2;前一种反应能垒较低。5-硝基-1-氢-四唑衍生物的热分解包括直接开环途径和质子转移途径[11];开环可以是由N1-N2键断裂开环,也可以是C5-N4键断裂开环;开环之后,两个途径都由另一个N-N 键断裂产生一个N2。密度泛函计算表明[12],FOX-7不是通过C-NO2键直接断裂分解,而是通过硝基-亚硝基重排机理分解。硝基-亚硝基重排反应的活化能计算值为247kJ/mol,接近FOX-7分解活化能的实测值242kJ/mol。这些研究表明,含能材料化学键断裂生成小分子碎片之前,有时会发生氢迁移、异构化或重排反应。
用量子化学研究含能材料分解机理时,通常只考虑单分子过程,即将含能材料当作气相体系。显然,凝聚态中的邻近分子间相互作用在很大程度上影响甚至改变含能材料的分解历程。DFT 计算气相TNAZ(1,3,3-三硝基氮杂环丁烷)分解时,其NNO2和C-NO2键断裂能分别为159kJ/mol和171kJ/mol,接近气相TNAZ分解活化能实验值164.7kJ/mol;其HONO消除反应活化能分别为183.9和188kJ/mol,与固相分解活化能194.8kJ/mol相近;由此推测气相分解主要是N-NO2和C-NO2键的断裂,而固相分解主要是HONO 的消除[13]。随后的计算进一步证实,TNAZ初始分解有NO2断裂、HONO消去和开环3个过程。NO2断裂的速率最大,但在固体中后两个过程也有重要作用,即固体中邻近分子间相互作用影响反应历程[14]。用DFT 计算NTO 二聚体分解过程得知,二聚体经历活化能为367kJ/mol的基元反应之后,在总反应的第2步即生成CO2[15];而在单分子分解反应中CO是终产物之一[16]。类似地,用DFT-B3LYP方法研究5-氨基四唑(5-ATZ)的分解过程时发现,虽然N2消去反应是单分子分解的主要反应途径,其氢键二聚体能显著地降低HN3消去反应的活化能,使HN3的消去反应优先进行,这与实验结果相一致[17]。
含能材料分解时产生高温,导致激发态反应的发生,有时甚至占主导地位。探讨激发态反应过程可揭示一些新的实验现象。例如,NO是RDX 激发态(经226~258nm 光子激发)分解时唯一观测到的小分子产物[18]。S1和S2态垂直激发能的计算值分别为5.59和6.1eV,表明RDX分子吸收226~258nm(5.5~4.8eV)光子后被激发至第一激发态。计算结果表明[19],其激发态分解须经历S1/S0圆锥交叉点(CI)的nitro-nitrite异构化非绝热过程。第2 垂直激发态S2,FC分解时,必然经历S2/S1圆锥交叉点(S2/S1)CI。即从S2态分解时,其最小能垒通道为S2(FC)→(S2/S1)CI→(S1/S0)CI→S0(nitrite)→NO 消去。此外,还发现2个协同的nitro-nitrite异构化过渡态,表明分解同时产生3个NO分子,与硝胺类分子激发态分解时NO信号强度正比于N-NO2数量的实验结果相符。图1为RDX激发态分解所产生的NO发生[A2Σ+ (ν′=0)←X2Π(ν″=0,1,2,3)]跃迁时的REMPI光谱,其转动和振动温度分别为20K 和1 800K[20]。计算结果与REMPI 光谱相吻合。高振动温度(1 800K)产物NO 的形成,表明激发态RDX 经势能面交叉点快速转变成基态,并沿基态势能面分解。HMX和CL-20的激发态分解与RDX 相似,也产生低转动温度及高振动温度的NO。与此相反,实验发现二甲硝胺经226nm 光子激发并分解后,产生高转动温度(120K)NO,CASSCF的计算结果预测了上述现象[20]。
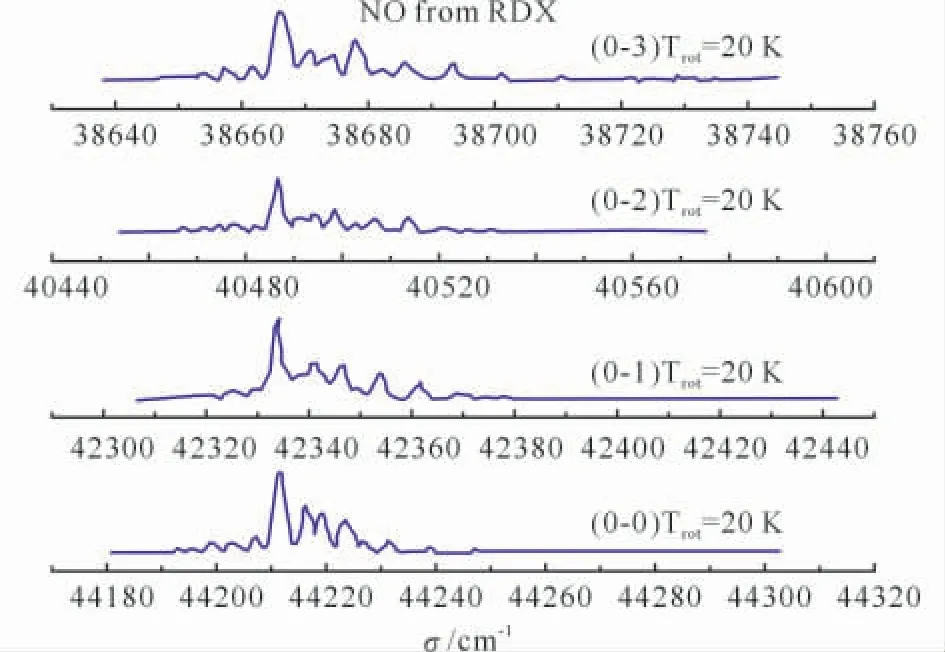
图1 RDX 激发态分解产物NO 的高分辨REMPI谱(转动和振动温度分别为20和1 800K)Fig.1 REMPI spectra of the NO product from excited electronic state decomposition of RDX(Spectra rotational and vibrational temperature are 20and 1800K,respectively)
需要指出的是,由于目前无法对含能材料反应势能面进行扫描,即无法确定势能面上所有的鞍点和能量极小点,导致不同文献对相同分子的分解机理报道不完全一致。当然,在少数情况下计算方法也导致结果的差别。故用量子化学方法探讨反应机理时,应注重不同计算方法之间的相互比较和印证。
含能材料的分解过程涉及温度压力变化和多分子多组分碰撞。量子化学无法完整揭示这些过程,需要借助分子动力学模拟进行补充和完善。后者不仅揭示温度和压力的影响,还给出中间产物、后续反应过程和各物种随时间的分布。对RDX 在高冲压下的分子动力学模拟表明,低压缩比时RDX较少分解,当压缩比大于40%时RDX 快速分解,并预测了中间产物(NO2,NO,HONO,OH)和最终产物(H2O,N2,CO,CO2)的分布规律[21]。基于量子力学多尺度分子反应动力学模拟研究TATB受到9 km/s冲击作用0.43ns及10km/s冲击作用0.2ns时,发现TATB形成富氮杂环分子簇,从而阻碍了TATB进一步分解[22]。
QC与MD 相结合,可揭示起爆或燃烧过程中的反应历程、决速步骤及活化能、催化机理、主次反应、产物分布、温度和压力等外界因素对反应过程的影响等。今后反应机理理论研究将更注重多分子和多组分体系、激发态过程以及界面反应。
3 感 度
20世纪70年代末,人们依据构效关系模型及其主要建模参数,相继提出以下撞击感度判据:氧平衡、激发态电荷梯度(ΔC*/l)、最小键级、静电势、活化能、键离解能和化学位移等。除早期的氧平衡判据外,其他感度判据均为量子化学参数,并且主要针对撞击感度而言。
早期炸药感度研究中,Kamlet等归纳了大量有机炸药实验数据,提出感度-氧平衡理论。经实验数据拟合,建立了根据炸药氧平衡计算撞击感度的公式[23]。显然,这一感度判据没有直接涉及含能材料的化学属性,特别是没有涉及其电子结构,无法揭示感度的本质。
随着理论研究的不断发展,发现仅考虑炸药分子内基团的特性和数目是不够的,必须深入到电子结构。Depluech等用半经验量子化学方法计算RNO2类典型炸药,发现基态电子结构和吸收能分布决定炸药的起爆性能[24],而这两个因素又影响到激发态的电子结构,并建议用ΔC*/l值(ΔC*是激发态下R-NO2键两端原子上净电荷之差,l为RNO2键的键长;两者的比值为激发态R-NO2键上的电荷梯度)作为硝基类炸药的撞击感度判据。这是从电子结构层次阐明炸药爆炸性能的开拓性工作。但对结构差别较大的炸药,其ΔC*/l值与感度的相关性不显著;另外并非任何外界作用都会使炸药的分子从基态跃迁到激发态。将电子激发过程的难易程度用于判断含能材料晶体的感度,发现撞击感度与前沿带隙相关,带隙越小撞击感度越大[25]。这一规律适用于离子型或分子型晶体同质异晶体及不同压力和掺杂条件[26]。新近对硝基苯类化合物的DFT 计算表明,感度主要依赖于LUMO 能级,其次与HOMO 能级有关[27]。
分子中最弱键的键级或双原子作用能与炸药的撞击感度或热安定性存在平行的递变关系,肖鹤鸣等用基态分子中最弱键的键级或该键连接的双原子相互作用能来判别同系物的撞击感度[28]。随后发现对引发键为C-NO2的多硝基芳香类炸药,其Mulliken键级与实测感度线性相关。但键级判定感度适用于共价型化合物,对于离子型化合物不适用,且只限于单键引发断裂过程。
DFT 计算表明,化学键离解能与相应静电势之间存在着关联[29]。Politzer等进一步证明,C-NO2键中点的静电势极值与C-NO2键的离解能近似成反比关系,因此用C-NO2键中点的静电势可以表征C-NO2键的强度,进而确定相应硝基化合物的感度[30]。对硝基化合物计算后发现,热解引发的反应活化能Ea与实验撞击感度存在线性关系:Ea越小,感度越大[31]。即用热解反应引发的活化能判断结构相似炸药的撞击感度[32]。热解反应引发的活化能动力学判据更有普遍性,但局限于分子结构和分解机理类似的化合物。
用DFT 方法对硝基芳香族炸药计算发现,感度的对数与炸药分子中引发键C-NO2的键离解能线性相关[33]。硝基化合物最弱键的键离解能与整个分子键离解能平均值之比(D/Ed,D为最弱键的键离解能,Ed为分子总的键离解能/键数目)可作为冲击感度的指标,该参数与16种重要爆炸物的冲击感度有显著的线性关系(R≥0.95);D/Ed值包含整个分子能量因素,在预测冲击感度方面有更好的表现[34]。
Zeman将多硝基化合物的撞击与电火花感度,爆炸与热分解的特征及其13C 及15N 的NMR 化学位移相关联[35]。此外,将核独立化学位移(NICS)与总能量的乘积作为描述符,得到一种新的芳香类炸药感度评估方法[36]。张朝阳等用硝基静电荷表示基团的吸电子能力,并将硝基电荷作为判断硝基芳香族化合物撞击感度的依据(即硝基电荷法)[37]。之后又发现分子中取代基的相互作用能(IE)可判断硝基化合物衍生物的稳定性,并成功地预测硝基苯和硝基苯胺类炸药的撞击感度[38]。除了用量子化学参数描述感度外,近年来分子动力学模拟也用于阐明感度与结构的关系。对TATB 的分子动力学模拟表明,当TATB 固体受到机械撞击时,其层状π结构不仅通过分子平面的滑移缓冲机械冲击作用,还可将冲击时的部分机械能转化为分子间作用能,从而减缓分子振动和热点形成[39],解释了TATB的钝感特性。
对单质和复合含能材料27种样品的实验研究表明,特性落高能(H50与落锤质量的乘积)的对数与热爆发活化能之间非常显著地线性相关[40]。这种适合于不同类型炸药的关系式,充分表明热爆发活化能与感度之间的本质关联。如何从理论上求得单质和复合炸药的热爆发活化能,建立既有明确物理意义、又同时适用于单质和复合含能材料的感度预测模型,对理论工作者提出了新的挑战。总之,影响感度的因素很多,仅当引爆过程主要受控于化学作用时,与化学活性相关的量子化学参数(最小键级、分解活化能、前沿分子轨道能隙、离去基团电荷等)才对感度起决定作用。这意味着感度预测模型的多样性及各自的局限性。
4 分子间相互作用及相容性
无论是由单组分还是多组分构成的含能材料,虽然分子组成和结构对其性能起决定作用,但各组分多聚体或混合组分之间的分子间相互作用,对它们的聚集状态、(液体)黏度、(固体)堆积方式和密度以及材料的多种性能(相容性、迁移性能等)也产生重要影响[41]。近10 余年来,量子化学方法已用于研究含能材料分子间的相互作用及其对含能材料性能的影响。对FOX-7二聚体和晶体的DFT 研究发现,最稳定二聚体的构型与晶体中分子堆积方式相似。说明结合能对该晶体中分子的排列方式起决定作用,还预示可按晶体结构较方便地找到某些分子的二聚或多聚体稳定构型[42]。在室温下由FOX-7单体生成最稳定二聚体的ΔG<0,即该二聚体可自发生成。在常温下结合能较弱的含能材料通常自发形成晶体,在很大程度上归因于分子间相互作用的协同效应[41]。
分子间相互作用的强弱从本质上决定了多组分体系相容性的大小。对于由分子间作用力结合而成的两组分体系,当A…A + B…B=2A…B 的ΔG<0,则A 与B 自发混合,即完全相容。通常A…B结合能大有利于两者之间的相容。在实际体系中,A 或B在混合前后均尽可能多地与邻近分子产生相互作用,并且A 与B分子的大小和形状各不相同。因此,直接由二聚体结合能值判断组分间的相容性并不具有普遍性。更为可靠的相容性判断方法是通过分子动力学模拟,得到溶解度参数和相互作用参数等。
从热力学角度来看,表征体系的相容性可用混合热ΔHm、混合熵ΔSm、溶解度参数δ及相互作用参数。其中溶解度参数较为简便,可根据溶解度参数差值(Δδ)预测高分子混合物之间的相容性。对于高分子体系,若分子间没有强极性基团或氢键作用,两种材料的Δδ只要满足cm-3)1/2两者就相容[43]。应用分子动力学(MD)和介观动力学(Meso-Dynamics)对固体推进剂中端羟基聚丁二烯(HTPB)与增塑剂癸二酸二辛酯(DOS)、硝化甘油(NG)的相容性模拟,得到等密度图、自由能密度和有序度参数等即可判断共混体系的相容性;MD 和介观模拟结果均表明,HTPB/DOS属于相容体系,DOS 在整个区域近似均匀分布,与HTPB 相容;而HTPB/NG 属于不相容体系,与实验结果一致[43]。
5 固体与界面性质
大多数含能材料在常温下是固相。与气相计算模型相比,固体模型包含邻近分子的相互影响。用固体模型可研究爆燃传播、晶格缺陷、机械能与热能转化、热点形成、振动态激发与键断裂、晶体能、弹性系数和膨胀系数等固体性质。碱金属叠氮盐晶体的计算结果表明,其前沿轨道均由叠氮根端位氮的原子轨道组成。由于碱金属离子对前沿空轨道的贡献极小,不利于电子从叠氮根向金属离子跃迁[44]。与此相反,叠氮化银和叠氮化铅晶体前沿占有的轨道主要由叠氮根端位氮的原子轨道组成,而前沿空轨道则主要由金属离子的原子轨道组成,并且带隙较小,这均有助于氮原子上电子直接向金属离子跃迁[45]。这可解释碱金属与过渡金属叠氮盐感度的差别。
对于固体计算,人们通常选用计算效率较高的DFT 方法。但DFT 法预测的色散力小于实测值,对分子间作用力较弱的分子晶体,计算结果存在较大误差。因此,理论化学家致力于对传统DFT 法进行改进。新近发展的DFT-D 方法能修正远程散色作用。用DFT-D 计算10 种典型含能材料分子晶体,其晶胞参数与实验结果的相对误差约2%或更少[46]。在常压下以及高压下的晶胞参数和体积随压力变化值与实验一致,其晶胞参数的最大误差为3.67%。不仅能较好地再现分子晶胞结构,还能准确预测晶胞中分子的相对位置和取向,为含能材料分子晶体计算提供更好的结果。
含能材料大多是混合体系,界面现象普遍存在。用DFT 共轭梯度近似(DFT-GGA)研究硝基甲烷和1,1-二胺基-2,2-二硝基乙烯在α-Al2O3(0001)表面上的行为[47],发现物理吸附的最稳定构型为分子面平行于Al2O3(0001)面。硝基甲烷分解的最低能垒(58.5kJ/mol)为H 原子消去;其C-N断裂能垒为158.8kJ/mol,均小于气相分解时的能垒。FOX-7在α-Al2O3(0001)表面上不发生分解。表明Al2O3表面氧化层虽有纯化作用,但对炸药分解有一定的催化能力[47]。用DFT-GGA 法研究硝胺分子在Al(111)表面的吸附表明[48]:既有物理吸附又有化学吸附。物理吸附时,硝基氧原子指向铝表面。化学吸附时,氧和氮原子与表面铝原子形成化学键,N-O 和N-N 键断裂,形成强的Al-O 和Al-N 键,铝表面可被硝基和氨基分解出的N 和O原子迅速氧化。DFT-GGA 法研究TNT 在铝(111)表面吸附结果表明,氧和铝原子间强烈的吸引作用使TNT 中N-O 键分解。TNT 邻位硝基的N-O 键比对位硝基的N-O 键易断裂。除了硝基的N-O 键断裂外,其他键未断裂,这有别于TNT 直接分解时C-NO2键优先断裂。可见与Al表面的作用改变了反应历程。含能材料在Al表面的分解过程,实际上是含铝炸药爆燃后期,Al表面氧化层破裂后发生的反应。因此,综合考虑金属氧化层及其厚度、氧化层高温破裂过程对含能材料吸附和分解的影响尤为重要。
固体和界面的一些性质,特别是涉及动态过程时,需要借助分子动力学模拟。用第一性原理分子动力学模拟RDX 在Al(111)表面的吸附和分解的结果表明,氧与铝的强吸引作用使RDX 的N-O和N-N 键断裂并产生NO;此外,RDX 环也发生断裂[49]。分子动力学模拟HMX 与石墨界面的弯折和滑移行为,得到表面结构和能量变化过程,表面弛豫和摩擦均对热点形成有贡献[50]。采用MD模拟键合剂对丁羟推进剂中端羟基聚丁二烯(HTPB)与Al/Al2O3之间界面的吸附能与力学性能的影响时发现[51]:键合剂在Al2O3晶面的吸附能高于HTPB在Al2O3晶面的吸附能,而在Al晶面的规律并不明显。Al/HTPB吸附界面加入键合剂TEA后,随其浓度的增加,体系弹性模量增大。对DOS、IPDI和HTPB组成的丁羟推进剂黏接体系的运动过程进行模拟[52],求得增塑剂DOS 在黏合剂体系(HTPB+IPDI)中的扩散系数,扩散系数随环境温度的升高而增大,随增塑剂含量的增大而减小,模拟结果与实验结果吻合,揭示了微观迁移现象。由于微观或介观模型的建立及模拟条件的选取常存在一定随意性,影响模拟结果的再现性,故特别强调模型应包含真实体系主要特征并且有统计意义。
6 极端条件下的性质
在实验技术难以实现的极端高压和高温下,用量子化学计算含能材料高压性质和MD 模拟其化学演变过程显得尤为重要。但经典MD不能对化学反应体系进行模拟,近10年发展的反应力场(ReaxFF)方法[53]和从头算CPMD方法为模拟含能材料高温高压反应过程提供了新的技术途径。ReaxFF反应力场模型依据键距与键级、键级与键能的关系,描述化学键断裂时体系能量变化,在原来的经典力场中加入键级参数及其修正项,比量子化学计算速度快万倍[53],可以模拟含数百万原子体系的反应过程[54]。
硝基甲烷在均匀受压和3个轴向受压时的量子化学计算结果表明[55],在静水压下,当体积压缩至原来的50%时,压力上升至50GPa,ΔEHOMO-LUMO减小了约0.6eV。最高占有轨道(HOMO)和最低空轨道(LUMO)同时增加,其差值却几乎随体积的变化单调下降,带隙减小从理论上可以认为感度在增加。在y方向上,带隙最多可减小1.4eV,比静水压时带隙下降值大,并且带隙呈非单调变化。x方向的应变引起的能隙变化与y方向的类似,只不过当V/V0接近50%时y方向带隙会引起陡降。当V/V0为65%~80%而z方向的能隙急剧下降。z方向压缩时,分子相互靠近时会导致一个分子的甲基与其邻近分子的硝基靠近,定域在硝基上HOMO和LUMO 的电子密度比其他类型的压缩形变更大,能隙下降得更快。高压所产生的y轴向张力,导致单胞内每个硝基甲烷分子均有1个C-H 键产生急剧伸展。随着y轴应变增加至50%,会导致这些键被进一步拉伸并引起质子消去。
HMX 和TATB 高温分解的分子动力学模拟表明,TATB经快速分解(30ps)产生大量多芳环为主的碳簇合物(总量的15%~30%),HMX 则产生小分子产物。HMX 在较低温度下易于分解,TATB分解速率比HMX 小一个数量级[54]。这可解释两种炸药分解过程实验现象的差别。分子模拟还揭示高温分解时各物种浓度变化。
对液体和固体硝基甲烷在2 000~3 000K、密度1.97g/cm3、时间200ps内的高温过程进行模拟[56],在3 000K 时分解起始步骤为分子间质子迁移,生成CH3NOOH 和CH2NO2。在较低的温度(2 500~2 000K)第一步反应则为异构化生成CH3ONO 的过程。同时在反应刚开始极短时间内可伴随分子内质子迁移生成CH2NOOH。作为放热过程的标志,H2O 的形成则是在这些过程之后。2000K 时液态硝基甲烷异构化生成亚硝酸酯,与从头算结果相一致。
采用MD 研究含能材料的分解过程,一般用ReaxFF力场。目前的ReaxFF参数通过拟合DFT计算结果得到。而DFT 方法低估了分子间的London色散力,导致晶体平衡体积偏高约10%~15%。最近通过对长程色散作用进行低梯度校正,得到ReaxFF-lg力场[57]。由室温下RDX 分子质心距离径向分布函数可知,ReaxFF-lg使分子模拟结果大为改进。预期这类修正的反应力场将对高温高压条件的含能材料研究发挥更有效的作用。
7 结束语
(1)随着量子化学和分子动力学模拟方法在含能材料领域的广泛应用,研究对象逐渐从分子结构简单体系扩展到复杂体系,并从单相扩展到多相、从体相扩展到界面。
(2)分子动力学模拟从平衡态模拟发展到施加外场的非平衡态模拟。尤其是介观层次的模拟不仅揭示纳米材料的特殊性质,还构筑了微观结构与宏观性质之间的桥梁。但介观体系涉及量子效应,如何改进量子化学方法,以实现介观层次的量子化学研究,对理论工作者提出了挑战。
(3)可以预测,量子化学和分子模拟对开发新型含能材料将发挥明显的、不可替代的作用。但是理论模拟不能代替实验,可靠的实验数据是检验模拟模型正确性的唯一依据。模拟和实验是相辅相成、缺一不可的研究手段,两者的结合将极大地促进含能材料的发展。
致谢:本综述的撰写得益于燃烧与爆炸技术重点实验室赵凤起研究员的鼓励、支持、并提出宝贵的修改意见,在此深表感谢!
[1]Repasky M P,Chandrasekhar J,Jorgensen W L.PDDG/PM3and PDDG/MNDO:Improved semiempirical methods[J].J Comput Chem,2002,23:1601-1622.
[2]Dorsett H,White A.Overview of molecular modelling and ab initio molecular orbital methods suitable for use with energetic materials[R].Salisbury:DSTO Aeronautical and Maritime Research Laboratory,2000.
[3]Keshavarz M H.Improved prediction of heats of sublimation of energetic compounds using their molecular structure[J].J Hazard Mater,2010,177(1-3):648-659.
[4]Gharagheizi F,Sattari M,Tirandazi B.Prediction of crystal lattice energy using enthalpy of sublimation:A group contribution-based model[J].Ind Eng Chem Res,2011,50:2482-2486.
[5]Kamlet M J,Jacobs S J.Chemistry of detonations.I.A simple method for calculating detonation properties of C-H-N-O explosives[J].J Chem Phys,1968,48:23-35.
[6]邱玲.氮杂环硝胺类高能量密度材料(HEDM)的分子设计[D].南京:南京理工大学,2007.
QIU Ling.Molecular design of high energy density materials(HEDM)-cyclic nitramines[D].Nanjing:Nanjing University of Science and Technology,2007.
[7]Ju X H,Li Y M,Xiao H M.Theoretical studies on the heats of formation and the interactions among the difluoroamino groups in polydifluoroaminocubanes[J].J Phys Chem A,2005,109:934-938.
[8]Fan X W,Ju X H,Xia Q Y,et al.Strain energies of cubane derivatives with different substituent groups[J].J Hazard Mater,2008,151(1):255-260.
[9]Fan X W,Ju X H.Theoretical studies on four-membered ring compounds with NF2,ONO2,N3,and NO2groups[J].J Comput Chem,2008,29:505-513.
[10]冯丽娜,张建国,张同来,等.氨基四唑化合物异构和分解反应的研究进展[J].含能材料,2009,17:113-118.
FENG Li-na,ZHANG Jian-guo,ZHANG Tong-lai,et al.Progress in the tautomerism and decomposition of amino-tetrazoles[J].Chin J Energetic Mater,2009,17:113-118.
[11]霍冀川,吴瑞荣,舒远杰,等.氮杂环类含能材料热分解研究进展[J].爆破,2007,24:21-25.
HUO Ji-chuan,WU Rui-rong,SHU Yuan-jie,et al.Investigative development of thermal decomposition of zaohetercycline energetic materials[J].Blasting,2007,24:21-25.
[12]王新锋,舒远杰.新型高能炸药热分解研究进展[J].化学研究与应用,2004,16(3):305-308.
WANG Xin-feng,SHU Yuan-jie.Studies on thermal decomposition of new high explosives[J].Chem Res Appl,2004,16(3):305-308.
[13]Alavi S,Reilly L M,Thompson D L.Theoretical predictions of the decomposition mechanism of 1,3,3-trinitroazetidine(TNAZ)[J].J Chem Phys,2003,119(16):8297-8304.
[14]Zhao Q,Zhang S,Li Q S.A direct ab initio dynamics study of the initial decomposition steps of gas phase 1,3,3-trinitroazetidine[J].Chem Phys Lett,2005,412:317-321.
[15]Kohno Y,Takahashi O,Saito K.Theoretical study of initial decomposition process of NTO dimer[J].Phys Chem Chem Phys,2001,3:2742-2746.
[16]Wang Y M,Chen C,Lin S T.Theoretical studies of the NTO unimolecular decomposition[J].J Mol Struct(THEOCHEM),1999,460(1):79-102.
[17]Kiselev V G,Gritsan N P.Theoretical study of the 5-aminotetrazole thermal decomposition[J].J Phys Chem A,2009,113(15):3677-3684.
[18]Guo Y Q,Greenfield M,Bernstein E R.On the excited electronic state dissociation of nitramine energetic materials and model systems[J].J Chem Phys,2007,127:154301.
[19]Bhattacharya A,Bernstein E R.Nonadiabatic decomposition of gas-phase RDX through conical intersections:An ONIOM-CASSCF study[J].J Phys Chem A,2011,115:4135-4147.
[20]Bhattacharya A,Guo Y Q,Bernstein E R.Nonadiabatic reaction of energetic molecules[J].Acc Chem Res,2010,43(12):1476-1485.
[21]Zhang L,Zybin S V,van Duin A C T,et al.Shock induced decomposition and sensitivity of energetic materials by ReaxFF molecular dynamics.Shock Compression of Condensed Matter[M].New York:American Institute of Physics,2006:585.
[22]Manaa M.R,Reed E J,Fried L E,Goldman N.Nitrogen-rich heterocycles as reactivity retardants in shocked insensitive explosives[J].J Am Chem Soc,2009,131:5483-5487.
[23]Kamlet M J,Adolph H G.The relationship of impact sensitivity with structure of organic high?explosives II.Polynitroaromatic explosives[J].Prop,Expl,Pyro,1979,4(2):30-34.
[24]Delpuech A,Cherville J.Relation entre la structure electronique et la sensibilitéau choc des explosifs secondaires nitrés.Critère moléculaire de sensibilitéII.Cas des esters nitriques[J].Prop,Expl,Pyro,1979,4(6):121-128.
[25]肖鹤鸣.金属叠氮化物的能带和电子结构[M].北京:科学出版社,1996.
[26]朱卫华,张效文,肖鹤鸣.高能晶体撞击感度理论研究:第一性原理带隙(ΔEg)判据[J],含能材料,2010,18(4):431-434.
ZHU Wei-hua,ZHANG Xiao-wen,XIAO He-ming.Theoretical studies of impact sensitivity of energetic crystals:First-principles band gap(ΔEg)criterion[J].Chin J Energetic Mater,2010,18(4):431-434.
[27]Türker L.Tunneling effect and impact sensitivity of certain explosives[J].J Hazard Mater,2009,169:819-823.
[28]肖鹤鸣.硝基化合物的分子轨道理论[M].北京:国防工业出版社,1993.
[29]March N H.The role of the bond midpoint electron density in homonuclear molecular binding[J].Int J Quantum Chem,1994,52(1):247-265.
[30]Politzer P,Murray J S.Relationships between dissociation energies and electrostatic potentials of C-NO2bonds:Applications to impact sensitivities[J].J Mol Struct(THEOCHEM),1996,376(1-3):419-424.
[31]Fan J F,Gu Z M,Xiao H M,et al.Theoretical study on pyrolysis and sensitivity of energetic compounds:(4)Nitro derivatives of phenols[J].J Phys Org Chem,1998,11(3):177-184.
[32]Chen Z X,Xiao H M.Impact sensitivity and activation energy of pyrolysis for tetrazole compounds[J].Int J Quantum Chem,2000,79:350-357.
[33]Rice B M,Sahu S,Owens F J.Density functional calculations of bond dissociation energies for NO2scission in some nitroaromatic molecules[J].J Mol Struct(THEOCHEM),2002,583:69-72.
[34]Mathieu D.Theoretical shock sensitivity index for explosives[J].J Phys Chem A,2012,116(7):1794-1800.[35]Zeman S.New Aspects of impact reactivity of polynitro compounds,Part III.Impact sensitivity as a function of the intermolecular interactions[J].Prop,Expl,Pyro,2003,28(6):301-307.
[36]杜军良,舒远杰,周阳.一种表征芳香族炸药撞击感度的简单方法[J].含能材料,2008,16(6):766-766.
DU Jun-liang,SHU Yuan-jie,ZHOU Yang.A simple method for predicting impact sensitivity of aromatic explosives[J].Chin J Energetic Mater,2008,16(6):766-766.
[37]Zhang C Y,Shu Y J,Huang Y G,et al.Investigation of correlation between impact sensitivities and nitro group charges in nitro compounds[J].J Phys Chem B,2005,109:8978-8982.
[38]Zhang C Y.Investigation on the correlation between the interaction energies of all substituted groups and the molecular stabilities of nitro compounds[J].J Phys Chem A,2006,110:14029-14035.
[39]Zhang C Y,Wang X C,Huang H.π-Stacked interac-tions in explosive crystals:Buffers against external mechanical stimuli[J].J Am Chem Soc,2008,130:8359-8365.
[40]刘子如,岳璞,任晓宁,等.热爆发活化能研究[J].火炸药学报,2011,34(6):58-63.
LIU Zi-ru,YUE Pu,REN Xiao-ning,et al.Investigation on activation energy of heat explosion[J].Chinese Journal of Explosives and Propellants,2011,34(6):58-63.
[41]肖鹤鸣,居学海.高能体系中的分子间相互作用[M].北京:科学出版社,2004.
[42]Ju X H,Xiao H M,Xia Q Y.A density functional theory investigation of 1,1-diamino-2,2-dinitroethylene dimers and crystal[J].J Chem Phys,2003,119(19):10247-10255.
[43]赵贵哲,冯益柏,付一政,等.端羟基聚丁二烯/增塑剂共混物相容性的分子动力学模拟和介观模拟[J].化学学报,2009,67(19):2233-2238.
ZHAO Gui-zhe,FENG Yi-bai,FU Yi-zheng,et al.Molecular dynamic simulations and mesoscopic dynamic simulations on the compatibility of HTPB/plasticizer blends[J].Acta Chim Sin,2009,67(19):2233-2238.
[44]Ju X H,Xiao H M,Guang F J.Periodic DFT approaches to crystalline alkali metal azides[J].Chin Sci Bull,2002,47:1180-1183.
[45]居学海,姬广富,邱玲.Cu+、Ag+叠氮盐晶体的周期性ab initio 计 算[J].高 等 学 校 化 学 学 报,2005,26
(11):2125-2127.JU Xue-hai,JI Guang-fu,QIU Ling,et al.Periodic ab initio calculations on Ag+and Cu+azides[J].Chem J Chin Univ,2005,26(11):2125-2127.
[46]Sorescu D C,Rice B M.Theoretical predictions of energetic molecular crystals at ambient and hydrostatic compression conditions using dispersion corrections to conventional density functionals(DFT-D)[J].J Phys Chem C,2010,114(14):6734-6748.
[47]Sorescu D C,Boatz J A,Thompson D L.First-principles calculations of the adsorption of nitromethane and 1,1-diamino-2,2-dinitroethylene (FOX-7)molecules on theα-Al2O3(0001)surface[J].J Phys Chem B,2005,109(4):1451-1463.
[48]Zhou S Q,Zhao F Q,Ju X H,et al.A density functional theory study of adsorption and decomposition of nitroamine molecules on the Al(111)surface[J].J Phys Chem C,2010,114(20):9390-9397.
[49]Umezawa N,Kalia R K,Nakano A,et al.1,3,5-Trinitro-1,3,5-triazine decomposition and chemisorption on Al(111)surface:First-principles molecular dynamics study[J].J Chem Phys,2007,126:234702.
[50]Long Y,Liu Y G,Nie F D,et al.Theoretical study of breaking and slipping processes for HMX/graphite interface[J].Appl Surf Sci,2012,258(7):2384-2392.
[51]焦东明,杨月诚,强洪夫,等.键合剂对HTPB 与Al/Al2O3之间界面作用的分子模拟[J].火炸药学报,2009,32(4):60-763
JIAO Dong-ming,YANG Yue-cheng,QIANG Hongfu,et al.Molecular simulation of effect of bonding agents on interface interaction for HTPB and Al/Al2O3[J].Chinese Journal of Explosives and Propellants,2009,32(4):60-763.
[52]李红霞,强洪夫,武文明.丁羟推进剂黏结体系中增塑剂迁移的分子模拟[J].火炸药学报,2008,31(5):74-78.
LI Hong-xia,QIANG Hong-fu,WU Wen-ming.Molecular simulation on plasticizer migration in the binding system of HTPB propellant[J].Chinese Journal of Explosives and Propellants,2008,31(5):74-78.
[53]van Duin A C T,Dasgupta S,Lorant F,et al.ReaxFF:A reactive force field for hydrocarbons[J].J Phys Chem A,2001,105:9396-9409.
[54]Zhang L,Zybin S V,van Duin A C T,et al.Carbon cluster formation during thermal decomposition of octahydro-1,3,5,7-tetranitro-1,3,5,7-tetrazocine and 1,3,5-Triamino-2,4,6-trinitrobenzene high explosives from ReaxFF reactive molecular dynamics simulations[J].J Phys Chem A,2009,113:10619-10640.
[55]Manaa M R,Fried L E,Reed E J.Explosive chemistry:Simulating the chemistry of energetic materials at extreme conditions[J].J Comput Aid Mol Des,2003,10(2):75-97.
[56]Han S P,van Duin A C T,Goddard III W A,et al.Thermal decomposition of condensed-Phase nitromethane from molecular dynamics from ReaxFF reactive dynamics[J].J Phys Chem B,2011,115:6534-6540.
[57]Liu L C,Liu Y,Zybin S V,et al.Correction of the ReaxFF reactive force field for London dispersion,with applications to the equations of state for energetic materials[J].J Phys Chem A,2011,115:11016-11022.
猜你喜欢
火炸药学报(2022年1期)2022-03-18
化工管理(2021年7期)2021-05-13
含能材料(2020年1期)2020-01-15
兵器装备工程学报(2017年10期)2017-11-14
火炸药学报(2014年5期)2014-03-20
火炸药学报(2014年5期)2014-03-20
火炸药学报(2014年5期)2014-03-20
火炸药学报(2014年5期)2014-03-20
火炸药学报(2014年5期)2014-03-20
火炸药学报(2014年5期)2014-03-20
