
4种天然纤维素在氢氧化钠/尿素/水体系中的溶解差异
2012-01-05 02:10:48邓海波李中石刘德桃
中国造纸学报 2012年3期
邓海波 李中石 吴 真 刘德桃 林 鹿 龙 柱
(1.江南大学生态纺织教育部重点实验室,江苏无锡,214122;2.华南理工大学制浆造纸工程国家重点实验室,广东广州,510640;3.淮阴师范学院江苏省生物质能与酶技术重点实验室,江苏淮安,223300)
近年来,随着石油资源的短缺,以可再生的生物质资源代替石化产品日益受到关注[1]。纤维素是地球上最丰富的可再生资源,来源极为广泛,因此开发和利用这种新型的环境友好型资源,是实现可持续发展的必然要求[2]。但是天然纤维素结晶度高,分子间及分子内存在大量氢键,因而难溶解、难融化,影响其应用[3]。
传统的纤维素溶解体系黏胶法还存在很多缺点,如释放有毒气体、破坏生态环境,铜氨法中铜和氨消耗量大,且难以回收利用,污染严重;氯化锂/二甲基乙基酰胺(LiCl/DMAc)体系由于成本高、操作复杂和不可回收循环利用等,目前还基本停留在实验室阶段;最近,N-甲基吗啉-N-氧化物(NMMO)、离子液体等新型绿色环保溶解体系备受关注,然而这些试剂价格昂贵[4]。文献[5-7]介绍了氢氧化钠/尿素/水溶解体系,利用这种新溶剂体系制备出新型纤维素膜、色谱柱填料、复合材料和纤维素衍生物等;并且以该体系为溶剂,以H2SO4和Na2SO4水溶液作为凝固剂,纺出了再生纤维素丝,这种再生纤维素丝性能类似于铜氨丝(bemberg)和天丝(lyocell纤维),染色性能却高于黏胶纤维[8-9]。该溶解体系工艺流程简短、生产过程无毒无害、溶剂可回收利用且价格低廉,因此具有良好的应用前景。
不同来源和种类的纤维素由于本身复杂的层次结构和超分子结构的差异,在这种溶解体系中的溶解机制也会有差别。因此,研究不同纤维素在该溶解体系中的溶解机制的差异、认识处理前后纤维素的变化规律,能够拓宽纤维素的应用领域,并推动纤维素工业的发展。本实验研究了棉短绒浆、竹浆、阔叶木浆和针叶木浆的天然纤维素在该溶解体系中的溶解差异。
1 实验
1.1 原料
棉短绒浆、竹浆、阔叶木浆、针叶木浆均为溶解浆(取自山东海龙化纤股份有限公司),棉短绒浆的纤维素含量大于99%,其他3种浆的纤维素含量均大于96%。
1.2 纤维素的溶解工艺
[4]的方法,在锥形瓶中加入100 mL氢氧化钠/尿素/水混合溶液,其中,NaOH质量分数7%、尿素质量分数12%,在-20℃条件下预冻2 h。取出后在室温下放置10~15 min,分别称取4种纤维素各4.00 g倒入锥形瓶,在10~15℃操作环境下,以2000 r/min的转速搅拌30 min,再将其放回冰柜,在-20℃下放置5 h,再在10~15℃操作环境下,以2000 r/min的转速搅拌30 min,即得到纤维素溶液,放在5℃下保存。
1.3 再生纤维素的制备
将制得的纤维素溶液分批倒入离心管中,以4000 r/min的转速离心10 min,以去除溶液中的气泡,然后在-80℃下冷冻24 h,再对其进行冷冻干燥处理,得到固体纤维素样品,即为再生纤维素。将所得样品用去离子水进行冲洗、抽滤处理,直到纤维素的pH值为7,并在40℃下真空干燥48 h,将得到的再生纤维素装入自封袋中备用。
1.4 聚合度的测定
按照 GB/T 5888—86的方法测定纤维素的聚合度。
1.5 原纤维素和再生纤维素的表征及分析
红外光谱分析:在Thermo Electron Corporation公司制造的NICOLET NEXUS 470红外光谱仪上进行红外光谱扫描,采用KBr压片方式制备试样。扫描速率0.2 cm/s,扫描32次,扫描范围400~ 4000 cm-1。
样品的X射线衍射分析在德国Bruker AXS公司的D8 Advance型X射线衍射仪上进行。实验条件为CuKα辐射源,管电压45 kV,电流40 mA,扫描范围2θ=5°~50°,扫描速率 4°/min,步长0.02°。
利用PeakFit软件进行分峰处理,并采用式(1)计算出纤维素结晶度[10]。式(1)中:Cr为结晶度(%);Sc为结晶峰的面积,一般用002面衍射峰2θ≈22.5°的峰强度表示;Sa为非结晶峰的面积,一般用002面衍射峰与101峰间2θ≈18°的峰强度表示。

2 结果与讨论
2.1 纤维素在氢氧化钠/尿素/水体系中的溶解差异
将制得的4种纤维素溶液分别在室温下以4000 r/min转速离心10 min后发现,棉短绒浆和竹浆的纤维素溶液为均一液相,溶液无色透明;而阔叶木浆和针叶木浆的纤维素溶液以同样条件处理后,出现了较为明显的分层现象(见图1)。由图1可以看出,针叶木浆和阔叶木浆的纤维素溶液的上层为透明清液,黏度较小;下层为半透明胶体状物质,黏度较大,说明这2种纤维素在该体系下并未完全溶解;同等条件下棉短绒浆和竹浆的纤维素的溶解度大于阔叶木浆和针叶木浆。
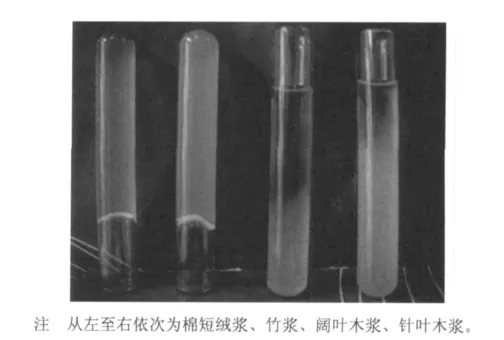
图1 离心后4种纤维素溶液
2.2 溶解前后纤维素聚合度的变化
通过铜氨溶液法测定原纤维素和处理后纤维素(再生纤维素)的聚合度(见图2)。由图2可知,处理后4种天然纤维素的聚合度都有不同程度的降低,棉短绒浆和竹浆的纤维素聚合度明显下降,从549和624降低至474和555,分别降低了13.6%和11.1%;而针叶木浆和阔叶木浆的纤维素聚合度降低不明显,从1067和1460降低至989和1419,只降低了7.3%和2.8%。这说明在该溶解体系中,各种天然纤维素在溶解过程中会发生一定程度的降解。但是降解程度与溶剂分子的可及度有关,聚合度较低的纤维素,其聚集态结构较为简单,溶剂分子的可及度高,纤维素的降解较为明显,故在该溶解体系的溶解过程中,棉短绒浆和竹浆的纤维素降解程度大于阔叶木浆和针叶木浆的纤维素。
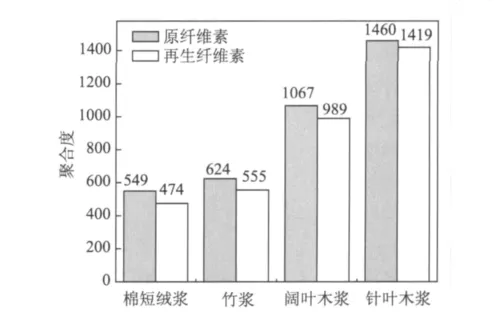
图2 纤维素聚合度的变化
2.3 处理前后纤维素分子结构特征的变化
图3为4种天然纤维素经氢氧化钠/尿素/水体系处理前后的红外光谱图。由图3可知,3340~3412 cm-1处宽峰为O—H的伸缩振动吸收所致,属于纤维素的特征吸收峰;2968、2900 cm-1附近的吸收峰归属于—CH2中C—H的伸缩振动吸收峰;1630 cm-1附近的吸收峰为纤维素吸收空气中的水所致;1431 cm-1和1316 cm-1附近的吸收峰为—CH2中C—H的摇摆振动;C—H的弯曲振动吸收峰出现在1373 cm-1和1281 cm-1附近;1201 cm-1处的吸收峰为葡萄糖环C6位的C—O—H的面内弯曲振动吸收峰;1237 cm-1为O—H的弯曲振动吸收峰;1158 cm-1和901 cm-1为糖苷键C—O—H的伸缩振动吸收峰;葡萄糖环的面内振动吸收在波数为1114、1061和1033 cm-1处的强吸收分别为 C3、C6位的 C—O吸收峰;在672、711 cm-1处的吸收为 C—O—H的面外弯曲吸收峰。
从图3可知,4种天然纤维素在处理前后的红外光谱图基本相同,而且没有新的吸收峰出现,但在强度上存在不同程度的差异,这说明在纤维素溶解过程中,没有在纤维素大分子上引入新的基团,只是对原有基团的振动强度产生影响。由此可以得出,纤维素在溶解过程中没有发生衍生化反应,氢氧化钠/尿素/水溶剂体系为非衍生化纤维素溶剂。
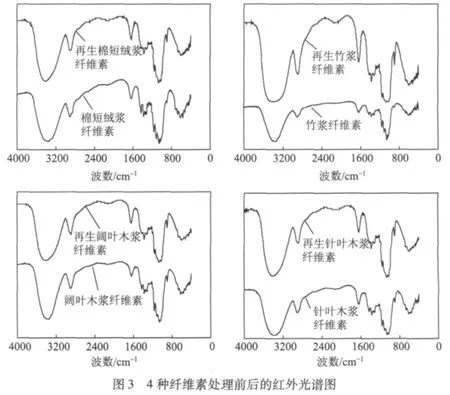
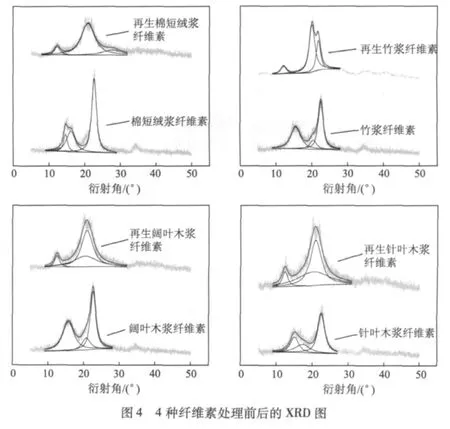
2.4 处理前后纤维素晶体结构的变化
根据X射线衍射峰可以方便地计算出纤维素的各种晶体参数,纤维素Ⅰ一般在 2θ=14.5°、16.5°、22.5°、34.5°附近,纤维素Ⅱ一般在 2θ=12°、20.2°、21.5°附近[11]。图4 为 4 种纤维素处理前后的XRD图,表1为4种纤维素处理前后的XRD结果分析。图4和表1的结果表明,4种纤维素样品经过处理后,纤维素Ⅰ的峰强基本消失,纤维素Ⅱ的峰开始出现,说明经过这种溶剂体系的处理,纤维素的晶体结构发生了转变,原有的Ⅰ型纤维素转变成Ⅱ型纤维素。从表1可以看出,棉短绒浆和竹浆的再生纤维素结晶度降低幅度比较大;结合再生前后纤维素聚合度的变化,说明由于阔叶木浆和针叶木浆的纤维素聚合度较大,氢氧化钠/尿素/水溶液体系不能有效破坏其结晶区,因而其溶解度小于棉短绒浆和竹浆的纤维素。
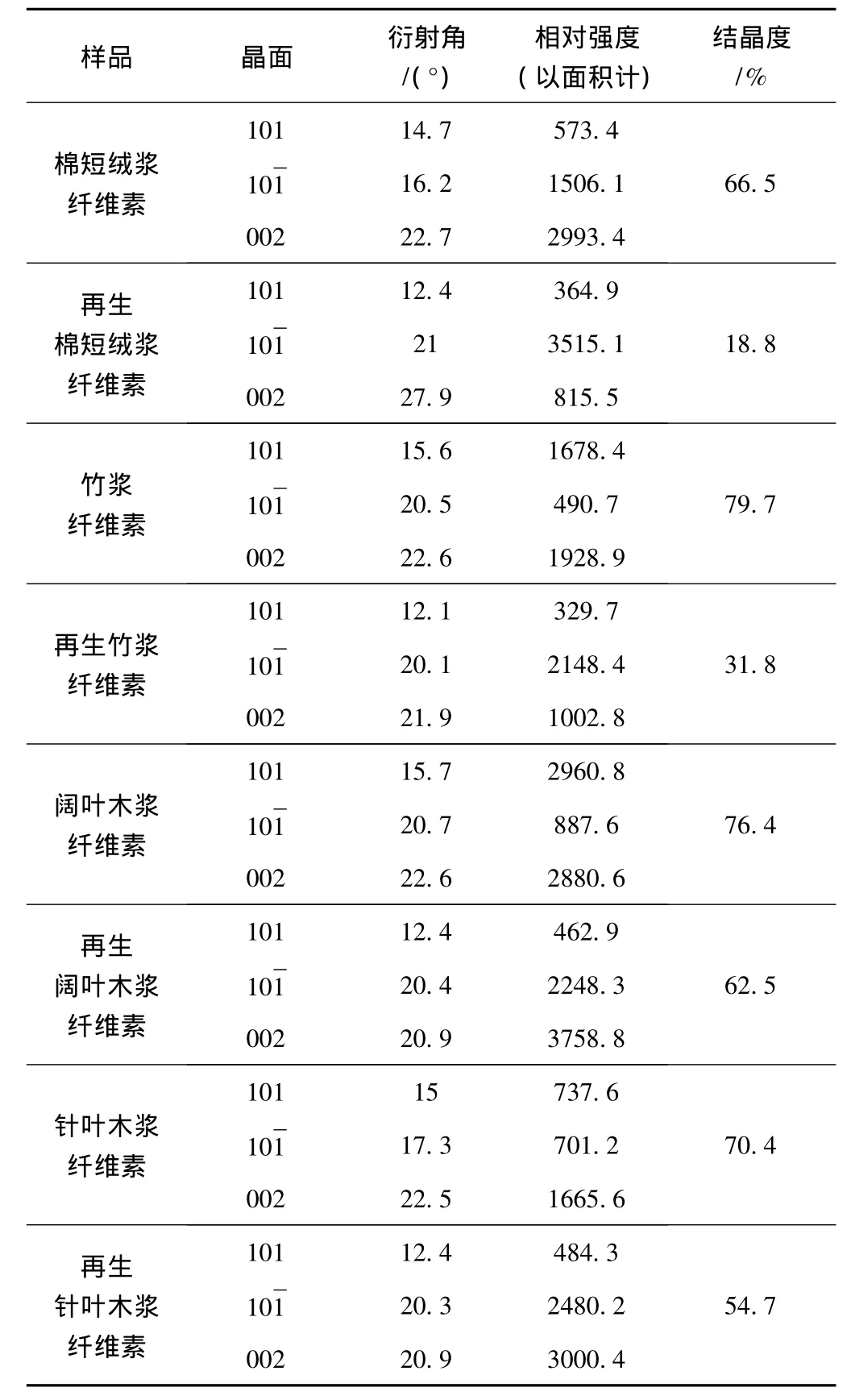
表1 4种纤维素溶液处理前后的XRD分析结果
3 结论
3.1 研究了棉短绒浆、竹浆、阔叶木浆和针叶木浆(均为溶解浆)4种天然纤维素在氢氧化钠/尿素/水体系中的溶解差异,棉短绒浆和竹浆的纤维素溶解后可形成均一的液相体系,而阔叶木浆和针叶木浆的纤维素溶液有分层现象,说明其没有完全溶解。相同条件下,棉短绒浆和竹浆的纤维素溶解度大于阔叶木浆和针叶木浆的纤维素。
3.2 氢氧化钠/尿素/水体系处理前后,4种纤维素的红外光谱图大体一致,在本实验的处理体系中,纤维素溶解过程中并未引入新的基团,说明氢氧化钠/尿素/水溶剂体系为非衍生化纤维素溶剂。处理后,纤维素的晶体结构由纤维素Ⅰ转变为纤维素Ⅱ。
3.3 由4种天然纤维素再生后得到的再生纤维素的聚合度和结晶度均有一定程度的降低,棉短绒浆和竹浆的纤维素聚合度从549和624降低至474和555,分别降低了13.6%和11.1%;而针叶木浆和阔叶木浆的纤维素聚合度降低不明显,从1067和1460降低至989和1419,分别只降低了7.3%和2.8%;棉短绒浆和竹浆再生后纤维素结晶度降低的幅度比较大,从66.5%和79.7%降低到18.8%和31.8%,分别降低了71.7%和60.1%;阔叶木浆和针叶木浆再生后纤维素结晶度降低的幅度较小,从76.4%和70.4%降 低 至 62.5% 和 54.7%, 分 别 降 低 了 18.2%和 22.3%。
3.4 天然纤维素在氢氧化钠/尿素/水溶液溶解体系中的溶解程度跟聚合度有关,由于阔叶木浆和针叶木浆的纤维素聚合度较大,该体系不能降低这2种纤维素的结晶度,因而其溶解度小于棉短绒浆和竹浆的纤维素。
参考文献
[1]Rostrup-Nielsen J R.Making Fuels from Biomass[J].Science,2005,308(5727):1421.
[2]Jiang X,Gu J,Tian X Z,et al.Modification of cellulose for high glucose generation[J].Bioresource Technology,2012,104:473.
[3]Cai J,Zhang L N,Liu S L,et al.Dynamic Self-assembly Induced Rapid Dissolution of Cellulose at Low Temperatures[J].Macromolecules,2008,41:9345.
[4]Cai J,Zhang L N.Rapid Dissolution of Cellulose in LiOH/Urea and NaOH/Urea Aqueous Solutions[J].Macromolecules Bioscience,2005,5(6):539.
[5]Cai J,Zhang L N,Zhou J,et al.Novel Fibers Prepared from Cellulose in NaOH/Urea Aqueous Solution[J].Macromolecular Rapid Communications,2004,25:1558.
[6]Mao Y,Zhou J,Cai J,et al.Effects of coagulants on porous structure of membranes prepared from cellulose in NaOH/urea aqueous solution[J].Journal of Membrane Science,2006,279:246.
[7]Zhang L N,Zhou J,Yang G,et al.Preparative fractionation of polysaccharides by columns packed with regenerated cellulose gels[J].Journal of Chromatography A,1998,816:131.
[8]Fink H P,Weigel P,Purz H J,et al.Structure formation of regenerated cellulose materials from NMMO-solutions[J].Progress in Polymer Science,2001,26:1473.
[9]Cai J,Zhang L N,Zhou J,et al.Multifilament Fibers based on Dissolution of Cellulose in NaOH/Urea Aqueous Solution:Structure and Properties[J].Advanced Materials,2007,19:821.
[10]吴翠玲,李新平,秦胜利,等.NMMO工艺纤维素膜结晶结构[J].江南大学学报:自然科学版,2006,5(3):352.
[11]Whitmore R E,Atalla R H.Factors influencing the regeneration of cellulose Ι from phosphoric acid[J].International Journal of Biological Macromolecules,1985(7):182.
猜你喜欢
食品与发酵工业(2021年14期)2021-08-02 12:47:08
生活用纸(2021年12期)2021-01-11 07:28:48
生活用纸(2016年7期)2017-01-19 07:36:47
生活用纸(2016年7期)2017-01-19 07:36:41
国际木业(2016年7期)2017-01-15 13:54:54
国际木业(2016年7期)2017-01-15 13:54:53
国际木业(2016年4期)2017-01-15 13:54:29
安徽化工(2016年5期)2016-02-27 08:25:04
浙江理工大学学报(自然科学版)(2015年7期)2015-03-01 02:54:21
天然产物研究与开发(2014年1期)2014-04-27 14:15:13
