
异恶唑衍生物缓蚀剂缓蚀性能的分子动力学模拟
2012-01-03 06:10孙霜青耿玉凤贾晓林郭爱玲胡松青
中国石油大学学报(自然科学版) 2012年1期
孙霜青,耿玉凤,贾晓林,郭爱玲,胡松青
(中国石油大学理学院,山东青岛266555)
异恶唑衍生物缓蚀剂缓蚀性能的分子动力学模拟
孙霜青,耿玉凤,贾晓林,郭爱玲,胡松青
(中国石油大学理学院,山东青岛266555)
采用分子动力学模拟方法研究盐酸环境下两种缓蚀剂(2-甲基-5-十二烷基异恶唑(A)和2-异丙基-5-十二烷基异恶唑(B))对低碳钢的缓蚀性能,并对其缓蚀机制进行分析。结果表明:在液相条件下,异恶唑缓蚀剂的极性头部优先吸附于金属表面,烷基碳链以一定角度指向溶剂,并且2-异丙基-5-十二烷基异恶唑的吸附强度大于2-甲基-5-十二烷基异恶唑;形成的致密缓蚀剂膜能有效阻碍腐蚀介质向金属表面扩散,达到减缓腐蚀的目的。
分子动力学;模拟;异恶唑;缓蚀剂;缓蚀机制
缓蚀剂分为有机缓蚀剂和无机缓蚀剂两种,有机缓蚀剂分子中含有N、O、S等易提供孤对电子的原子或不饱和键的活性基团[1],可在金属表面形成稳定的吸附膜,阻止腐蚀介质与金属表面接触。分子动力学(MD)方法不考虑电子的运动情况,把原子作为最小单元,已成为从分子水平上理解化学过程的一种有效手段[2-6],运用分子模拟方法[7-8]研究缓蚀剂分子在金属表面的吸附行为,通过计算体系中粒子的运动状态随时间的演化,可全面、真实地解释缓蚀剂的缓蚀机制。异恶唑[9-11]通常被用于合成对生物体有利的产品,Ali等[12]首次采用失重法和电化学方法等实验手段研究了异恶唑衍生物抑制盐酸对低碳钢腐蚀的缓蚀性能,并取得了良好的缓蚀效果。笔者以2-甲基-5-十二烷基异恶唑和2-异丙基-5-十二烷基异恶唑两种缓蚀剂[13]为研究对象,采用分子动力学模拟方法,研究这两种缓蚀剂分子在金属表面的吸附行为以及成膜特性,并深入分析其缓蚀机制。
1 计算方法
2-甲基-5-十二烷基异恶唑(A)和2-异丙基-5-十二烷基异恶唑(B)两种缓蚀剂的分子结构和缓蚀效率见表1。
缓蚀剂分子A和B的初始构型均由MaterialsStudio 4.0(Accelrys Software Inc.公司产品)软件包中的Visualizer模块构建。采用分子动力学模拟方法研究缓蚀剂分子与铁界面的相互作用。模拟过程中采用周期性边界条件,计算模型选用三层结构模型,构建过程如下:首先,构建金属表面,选取Fe(001)[14]晶面中的12层Fe原子,体系为3.153 nm×3.153 nm× 1.577 nm,共计1452个原子,模拟过程中冻结金属表面体系中的所有原子;其次,利用Amorphous Cell模块构建两种溶剂层,一种用于研究单分子的吸附行为,包含1 000个水分子和1个异恶唑衍生物分子,另一种用于研究多分子的吸附成膜行为,包含1 000个水分子和16个异恶唑衍生物分子;最后,通过Amorphous Cell模块构建包含500个水分子的溶剂层,模拟过程中冻结该层中所有原子的坐标。

表1 两种缓蚀剂的分子结构和缓蚀效率Table 1 Conformations and experimental inhibition efficiencies for two inhibitors
模拟采用Materials Studio 4.0软件包完成,利用Visualizer模块构建A和B的分子模型,力场为COMPASS[15]力场,通过 Discover模块下的 Smart Minimizer方法对体系进行优化,采用正则系综(NVT)[16]进行分子动力学模拟。模拟温度为333 K,采用Andersen[17]恒温器进行温度控制。各分子起始速度由Maxwell-Boltzmann分布随机产生。利用周期性边界条件和时间平均等效于系综平均等基本假设,运用Velocity Verlet算法[18]求解牛顿运动方程。范德华和库仑相互作用采用Charge Group方法[19]计算。截断半径为1.5 nm(spline width:0.10 nm,buffer width:0.05 nm),截断距离之外的分子间相互作用按平均密度近似方法进行校正。模拟时间为1000 ps,步长为1.0 fs,每1000 fs记录一次体系的轨迹信息。
缓蚀剂分子的结构及相关参数均在体系达到平衡后提取。体系是否达到平衡可由温度和能量判据判定。图1为缓蚀剂分子B动力学模拟过程中体系的能量和温度随时间的演化曲线(A分子具有相似的能量和温度分布曲线)。由图1可见,模拟500 ps后温度曲线波动在(333±10)K,能量涨落在0.3 %左右,表明体系确已达到平衡,因此选取后300 ps进行取样,计算相应缓蚀剂分子吸附构型结构参数和相关能量的统计平均值。
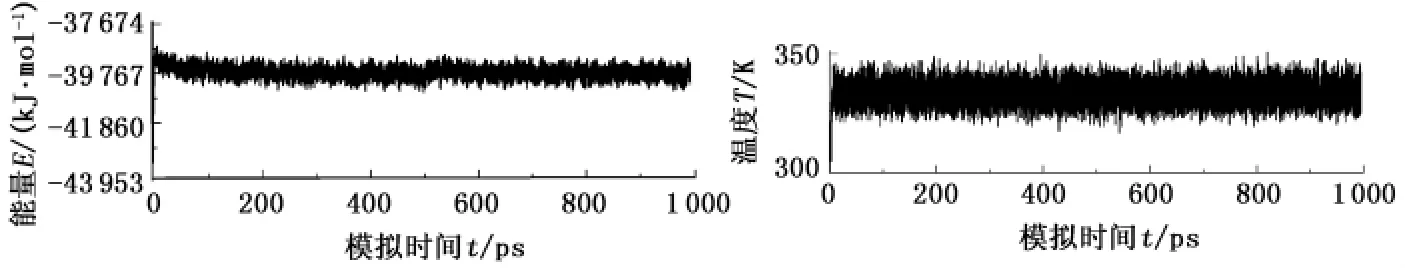
图1 2-异丙基-5-十二烷基异恶唑在Fe(001)表面吸附的能量和温度波动曲线Fig.1 Energy and temperature fluctuation curves of 2-isopropyl-5-dodecylisoxazolidine(B)on Fe(001)surface
缓蚀剂分子在Fe(001)表面的吸附能可由如下公式[20]计算得出:
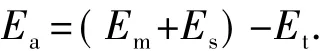
其中,Ea是缓蚀剂在金属表面的吸附能;Em是孤立缓蚀剂分子的能量;Es是未吸附缓蚀剂时金属表面的能量;Et是缓蚀剂分子和金属表面体系的总能量。能量的单位为千焦/摩尔。
2 结果分析
2.1 单分子吸附的分子动力学模拟
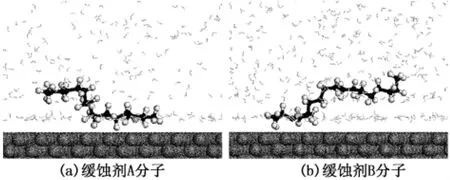
图2 液相中A和B分子在Fe(001)表面的平衡吸附构型Fig.2 Equilibrium structures of inhibitors A,B adsorbed on Fe(001)surface in liquid phase
液相条件下缓蚀剂分子在Fe(001)表面的平衡吸附构型见图2。可以看出,两种缓蚀剂分子的极性头部(异恶唑环)由于较强的化学反应活性总比烷基链优先吸附于Fe(001)表面,并且与缓蚀剂分子的起始位置无关,而烷基长链在溶剂水的作用下发生扭转并偏离金属表面以一定倾角指向溶剂。体系达到平衡后,缓蚀剂分子在其平衡位置附近作微小的振动,这种吸附特性既有利于缓蚀剂分子通过极性头基在Fe表面上形成稳定吸附,同时也有利于烷基碳链在金属表面自组装成疏水保护膜,阻碍与腐蚀反应有关的电荷和物质的转移,起到提高缓蚀性能的作用。
为细致描述缓蚀剂在金属表面的吸附现象,计算了体系平衡后异恶唑环在金属表面的吸附角及异恶唑环的质心与金属表面的距离,其统计平均值见表2。由表2可以看出,缓蚀剂A和B异恶唑环的吸附角(θ)分别为13.27°和11.07°,异恶唑环的质心与表面的距离分别为0.321和0.288 nm,表明缓蚀剂分子的极性头部具有平行吸附的趋势,并且缓蚀剂B较A有更强的吸附特性。

表2 A和B两种缓蚀剂分子液相条件下吸附角、距离和吸附能的统计平均值Table 2 Statistic average values of adsorption angles,distances and adsorption energies in solvent for two inhibitors A and B
缓蚀剂分子与金属表面之间吸附能能够在一定程度上反映缓蚀性能的强弱,其绝对值越大,表明缓蚀剂与金属表面的结合越稳定。由表2可知:两种异恶唑衍生物缓蚀剂分子的吸附能数据均为负值,表明异恶唑衍生物缓蚀剂在Fe(001)表面的吸附是体系放出热量、自由能降低的过程,即体系为稳定状态;此外,异恶唑衍生物缓蚀剂分子吸附能的绝对值分别为200.66和341.41 kJ/mol,远远大于水分子在Fe(001)表面的吸附能(-23.40 kJ/mol)[21],说明两种缓蚀剂分子均可驱替水分子而稳定吸附在Fe(001)表面;缓蚀剂B在Fe(001)表面吸附能的绝对值大于A,表明缓蚀剂B在Fe表面的吸附稳定性强于A,其缓蚀性能好,这与实验结果相吻合。
2.2 多个缓蚀剂在金属表面的吸附成膜
界面型缓蚀剂缓蚀性能的优劣,关键在于能否在金属表面形成高覆盖度且稳定的保护膜,能否有效阻碍与腐蚀反应有关的电荷或物质向金属表面迁移扩散。为进一步描述两种异恶唑缓蚀剂分子的吸附成膜特性,考察多个缓蚀剂在Fe(001)表面的吸附行为。图3为两种缓蚀剂在Fe(001)表面吸附成膜的平衡构型。
分子头基之间存在较强烈的相互作用,导致分子头基之间相互牵制而难以平行吸附,分子头基的这种吸附可减小单个缓蚀剂分子在金属表面的占有面积,有利于更多的分子吸附在金属表面,从而增强膜的致密性。此外,在溶剂的影响下缓蚀剂烷基碳链远离Fe表面且相互交织形成疏水的保护膜,可有效阻碍水分子向金属表面扩散,抑制腐蚀过程。图3 (b)中疏水烷基链的交织程度较(a)强,且缓蚀剂膜B驱替水分子的能力强于A,而两种缓蚀剂分子具有相同的烷基链(链上C原子数均为12),这表明分子头基上的官能团对交织成膜具有很大的影响,其活性越强,缓蚀剂构成膜驱替腐蚀介质的能力越强,膜的致密性也越好。
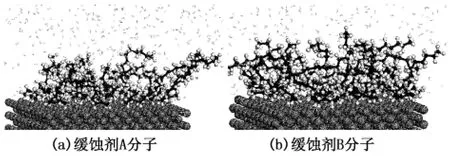
图3 缓蚀剂分子在Fe(001)表面吸附成膜的平衡构型Fig.3 Equilibrium structures of inhibitor monolayer on Fe(001)
为定量描述缓蚀剂膜对溶剂水分子的阻碍性能,分别计算了添加两种缓蚀剂前、后水分子在Fe(001)表面的数密度(在Fe表面垂直方向上距离表面不同高度处水分子的数量)随高度的分布曲线,结果见图4。
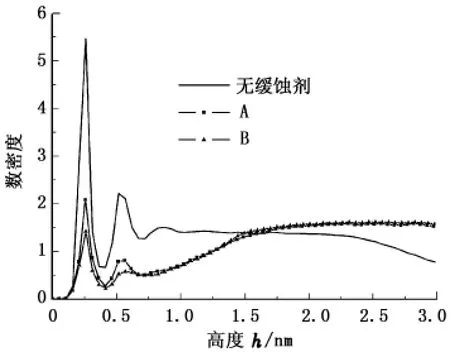
图4 金属表面水分子的数密度随高度的分布Fig.4 Number density profiles for water molecules on metal surface
从图4可以看出:添加缓蚀剂前、后水分子密度曲线的第一峰峰值均出现在距表面高度0.258 nm处,表明缓蚀剂膜与金属表面之间存在少量水分子,缓蚀剂分子未能完全驱替金属表面的溶剂水;与无缓蚀剂时相比,添加缓蚀剂A后水分子的密度峰峰值由5.465下降至2.08,而添加缓蚀剂B后水分子的密度峰峰值下降至1.42,可推断出,缓蚀剂分子吸附在Fe(001)表面,能有效驱替表面附近溶剂层中的水分子,并且缓蚀剂B驱替水分子的能力较A强。添加缓蚀剂后,水分子数密度在距 Fe表面0.412~0.716 nm的区间内明显下降,表明具有疏水性能的烷基长链团聚,排斥该处液相中的水分子使之远离金属表面。由此可知,在溶液中添加缓蚀剂后,其极性头基吸附在金属表面,非极性烷基链形成一层疏水膜,可以有效阻断溶剂水、氯离子和氧等腐蚀性粒子与金属表面的接触,从而达到阻碍或减缓腐蚀的效果。
3 结论
(1)异恶唑衍生物缓蚀剂的极性头部优先吸附在金属表面,而烷基碳链在溶剂水的作用下背离金属表面,以一定的倾角指向溶剂,并且缓蚀剂B的吸附稳定性强于A。
(2)多个缓蚀剂分子在金属表面形成致密的缓蚀剂膜,能有效阻碍水分子向金属表面扩散。缓蚀剂B排斥水的能力强于A,缓蚀剂B的缓蚀性能优于A。
[1]张天胜,张浩,高红.缓蚀剂[M].北京:化学工业出版社,2008.
[2]郭向丹,黄世萍,滕加伟,等.水在NanZSM-5型分子筛中的吸附的研究:分子模拟[J].物理化学学报,2006,22(3):270-274.
GUO Xiang-dan,HUANG Shi-ping,TENG Jia-wei,et al.Study on the adsorption of water on NanZSM-5 type zeolite:molecular simulation[J].Acta Physico-Chimica Sinica,2006,22(3):270-274.
[3]UNGERERP,NIETO-DRAGHI C,ROUSSEAU B,et al.Molecular simulation of the thermophysical properties of fluids:from understanding toward quantitative predictions[J].Journal of Mol Liq,2007,134(1/3):71-89.
[4]SRIVASTAVA P,CHAPMANW G,LAIBINISP E.Molecular dynamics simulation of oxygen transport through ω-alkoxy-n-alkanethiolate self-assembled monolayers on gold and copper[J].Langmuir,2009,25(5):2689-2695.
[5]侯廷军,朱丽荔,徐筱杰,等.MCM-22型分子筛中苯分子吸附行为的分子动力学模拟[J].物理化学学报,2000,16(8):701-707.
HOU Ting-jun,ZHU Li-li,XU Xiao-jie,et al.Diffusion of benzene in MCM-22 zeolite:a molecular dynamics study[J].Acta Physico-Chimica Sinica,2000,16(8): 701-707.
[6]韩振为,廖川,周薇.分子动力学模拟聚赖氨酸在晶格界面上的吸附[J].计算机与应用化学,2007,24 (5):703-708.
HANZhen-wei,LIAO Chuan,ZHOU Wei.Molecular dynamics simulation of adsorption of PLL(poly-lysine) on crystal lattice interfaces[J].Compu and Appl Chem,2007,24(5):703-708.
[7]张军,于维钊,燕友果,等.咪唑啉缓蚀剂在 Fe (001)表面吸附行为的分子动力学模拟[J].物理化学学报,2010,26(5):1385-1390.
ZHANG Jun,YU Wei-zhao,YANYou-guo,et al.Molecular dynamics simulation of the adsorption behavior of imidazoline corrosion inhibitors on Fe(001)surface[J].Acta Physico-Chimica Sinica,2010,26(5):1385-1390.
[8]张曙光,王风云,雷武.水溶性聚合物与硬石膏晶体相互作用的分子动力学模拟[J].化学学报,2007,65 (20):2249-2256.
ZHANG Shu-guang,WANG Feng-yun,LEI Wu.Molecular dynamics simulaton of interaction between water-soluble polymers and anhydrite crystal[J].Acta Chim Sin,2007,65(20):2249-2256.
[9]张军,李中谱,赵卫民,等.咪唑啉缓蚀剂缓释性能的理论研究[J].石油学报:石油加工,2008,24(5): 598-604.
ZHANG Jun,LI Zhong-pu,ZHAO Wei-min,et al.Theoretical study on corrosion inhibition performance of imidazoline inhibitors[J].Acta Petrolei Sinica(Petroleum Processing Section),2008,24(5):598-604.
[10]王业飞,由庆,赵福麟.一种新型咪唑啉复配缓蚀剂对A3钢在饱和CO2盐水溶液中的缓蚀性能[J].石油学报:石油加工,2006,22(3):74-78.
WANG Ye-fei,YOU Qing,ZHAO Fu-lin.Inhibition properties of a novel imidazoine complex for A3 steel in salt water saturated by CO2[J].Acta Petrolei Sinica (PetroleumProcessing Section),2006,22(3):74-78.
[11]CRUJZ J,MARTINEZ-AGUILERALLMR,SALCEDO R,et al.Reactivity properties of derivatives of 2-imidazoline:an abinitio DFT study[J].Int JQuantum Chem,2001,85(4/5):546-556.
[12]ALI SA,SAEED MT,RAHMANSU.The isoxazolidines:a new class of corrosion inhibitors of mild steel in acidic medium[J].Corros Sci,2003,45(2):253-266.
[13]ALI SA,EL-SHAREEF A M,AL-GHAMDI RF,et al.The isoxazolidines:the effects of steric factor and hydrophobic chain length on the corrosion inhibition of mild steel in acidic medium[J].Corrosion Science,2005,47(11):2659-2678.
[14]RAMACHANDRANS,TSAI B L,BLANCO M,et al.Self-assembled monolayer mechanism for corrosion inhibition of iron by imidazolines[J].Langmuir,1996,12 (26):6419-6428.
[15]SUNH.Compass:an ab initio force-field optimized for condensed-phase applicationss overview with details on alkane and benzene compounds[J].Phys Chem B,1998,102(38):7338.
[16]HEERMANND W.Computer simulation methods in the theoreticalphysics[M].Beijing:Peking University Press,1996.
[17]ANDERSENH C.Molecular dynamics simulations at constant pressure and/or temperature[J].Chem Phys,1980,72(4):2384-2393.
[18]吴兴惠,项金钟.现代材料计算与设计教程[M].北京:电子工业出版社,2002.
[19]LEACH A P.Molecular modeling:principles and application[M].England:Person Education Limited,2001.
[20]KORNHERRA,TORTSCHANOFF A,ERWINP C.Modelling of aqueous solvation of eosin Y at the rutile TiO2(110)/water interface[J].Chem Phys Lett,2006,430(4/6):375-379.
[21]胡松青,胡建春,高元军,等.月桂基咪唑啉对Q235钢的缓蚀吸附作用[J].化工学报,2011,62 (1):147-155.
HU Song-qing,HU Jian-chun,GAO Yuan-jun,et al.Corrosion inhibition and adsorption of lauryl-imidazolines for Q235 steel[J].CIESCJournal,2011,62 (1):147-155.
Molecular dyanmics simulation of corrosion inhibition performance of isoxazolidines derivatives inhibitors
SUNShuang-qing,GENG Yu-feng,JIA Xiao-lin,GUO Ai-ling,HU Song-qing
(College of Science in China University of Petroleum,Qingdao266555,China)
The corrosion resisting properties of two corrosion inhibitors including 2-methyl-5-dodecylisoxazolidine(A)and 2-isopropyl-5-dodecylisoxazolidine(B)in HCl on mild steel were theoretically evaluated using molecular dynamics simulations,and the corrosion inhibition mechanism was analyzed.The results show that the polarity head group of isoxazolidine corrosion inhibitors priority adsorbs on the metal surface under the condition of liquid phase,while the alkyl chain stays in the water phase with distortion.And the adsorption intensity of inhibitor B is greater than that of inhibitor A.The inhibitor molecules could form a dense membrane,which could efficiently prevent the corrosive media from diffusing to the metal surface so as to slow down or even check the corrosion process.
molecular dynamics;simulation;isoxazolidines;corrosion inhibitor;corrosion inhibition mechanism
O 641;TG 174.42
A
10.3969/j.issn.1673-5005.2012.01.027
1673-5005(2012)01-0154-04
2011-09-12
中石化普光气田缓蚀剂技术研究项目(309003);中国石油大学(华东)研究生创新基金项目(S10-30)
孙霜青(1981-),男(汉族),河南新乡人,讲师,博士,研究方向为防腐蚀工程。
(编辑 刘为清)
猜你喜欢
大电机技术(2022年3期)2022-08-06
化工设计通讯(2022年5期)2022-05-25
河南化工(2022年2期)2022-03-21
石油化工技术与经济(2021年6期)2022-01-18
成都信息工程大学学报(2021年4期)2021-11-22
化工技术与开发(2020年11期)2020-11-26
当代化工(2019年1期)2019-12-12
山东工业技术(2018年16期)2018-09-26
造纸化学品(2017年1期)2017-01-21
少年科学(2015年7期)2015-08-13
