
链霉菌H197 mtg基因毕赤酵母表达载体的构建与鉴定
2012-01-03 08:11:09何冬兰王亚南邵坤彦
中南民族大学学报(自然科学版) 2012年1期
何冬兰,王亚南,邵坤彦,叶 程
(中南民族大学 生命科学学院, 武汉 430074)
谷氨酰胺转胺酶(TGase或TG)[1]在食品加工中具有一定的功能特性[2-4]. 20世纪90年代末,Ando[5]等首次发现Strepovertilliumsp.s-8112、S.mobaraense等具有产生微生物谷氨酰胺转胺酶(MTG)的能力,并首次用发酵法生产得到了MTG. 近2年, 有研究采用离子束诱变、紫外线及He-Ne激光复合诱变的方法对谷氨酰胺转胺酶菌株进行选育研究[6,7],效果均不太明显.这些方法对实验条件要求高,不适合工业化生产.利用基因工程的方法将MTG转入到原核生物中进行表达从一定程度上解决了MTG生产成本过高,产量不能满足食品工业的需求等问题.但原核生物对外源蛋白的表达存在一定的缺陷,需进行复性方可获得具有生物活性的目的蛋白.毕赤酵母菌是真核生物,能对表达蛋白进行加工[8-10],因此将MTG基因转入毕赤酵母中进行表达直接产生有活性的目的蛋白.目前国内外几乎未见将谷氨酰胺转胺酶转入真核生物中的研究. 本研究选用巴斯德毕赤酵母表达载体pPIC3.5k进行重组质粒的构建,将MTG基因插入pPIC3.5k中,转入至毕赤酵母菌中,下一步将进行载体的表达研究,以期待获得能在毕赤酵母中表达的目的产物,由此用于解决工业生产中蛋白复性的麻烦.
1 材料与方法
1.1 材料
1.1.1 菌种和质粒
毕赤酵母表达载体pPIC3.5k为Invitrogen公司产品;实验菌株E.coliDH5a和链霉菌Streptomycessp.H197为本实验室保存.
1.1.2 主要试剂
ExTaq酶、限制性内切酶EcoRI、BamHI和4 DNA连接酶均购自Takara公司;DNA Marker购自BBI公司;凝胶回收试剂盒购自Axygen公司.其他试剂均为进口或国产分析纯.
1.1.3 引物
根据Streptomycessp. H197基因序列,设计如下带酶切位点的上游引物和下游引物:
上游:BamHI CGTGGATCCGCCAGCGGCGGC
GACGGGGAAA;
下游:EcoRI CCGGAATTCTTACGGCCAGCCC
TGCTT, 引物合成由上海赛百盛基因技术有限公司完成.
重组质粒PCR鉴定引物合成,引物序列如下:
5′AOX1Primer GACTGGTTCCAATTGACAAGC;3′AOX1 Primer GCAAATGGCATTCTGACATCC,引物合成由上海赛百盛基因技术有限公司完成.
1.2 方法
1.2.1Streptomycessp. H197基因组DNA的提取
挑取4℃平板保存的单菌落接种于20mL液体培养基中[11],在30℃、200r/min条件下培养2~3d,采用改进的微波法提取总基因组[12]. 将提取的基因组用0.8%琼脂糖凝胶电泳检测,保存于-20℃冰箱.
1.2.2 PCR法对目的基因DNA的扩增
将提取的总基因组做为模板进行PCR扩增[13,14],PCR体系为25μL.

表1 PCR扩增体系
反应在Biometra PCR仪上进行,程序设定为95℃预变性5min;94℃ 1min,58℃ 1min,72℃ 90s,共35个循环;72℃ 10min,10℃保温30min.
PCR产物0.8%琼脂糖凝胶电泳检测,并用回收试剂盒将目的片段回收.
1.2.3 毕赤酵母胞内表达载体pPIC3.5k的提取
将pPIC3.5k/DH5α大量培养,采用碱裂解法取质粒,利用0.8%琼脂糖凝胶电泳检测,-20℃保存.
1.2.4 PIC3.5k/MTG酵母表达载体的构建
对凝胶回收的MTG基因片段和pPIC3.5k质粒分别进行BamHI和EcoRI双酶切,酶切产物电泳进行凝胶回收纯化获得具有粘性末端的质粒和目的片段,T4DNA连接酶连接,CaCl2法转化入感受态E.coliDH5α,涂布于含Amp的低盐LB平板进行筛选,选择重组菌落进行质粒小量制备[15-17].
1.2.5 重组表达质粒的PCR和酶切鉴定
将重组表达质粒进行PCR鉴定[18],选用目的片段的上游和下游引物鉴定目的片段的大小,选用目的片段的上游引物和3′AOX1确定目的片段插入的方向是否正确.
将重组质粒进行BamHI和EcoRI双酶切验证.
2 结果及分析
2.1 MTG基因的PCR扩增及其回收
由Streptomycessp. H197菌种提取的总基因组经过电泳检测(见图1)于23kb处可见明亮的总DNA带,说明提取效果很好.以Streptomycessp. H197菌株的总DNA为模板, 以设计的上游引物和下游引物扩增出约1.2kb的谷氨酰胺酶基因片段(见图2), 将MTG基因扩增体系扩大进行PCR扩增,经电泳后切胶回收,利用DNA凝胶回收试剂盒(小量DNA片段快速胶回收试剂盒)对目的DNA片段进行回收,并电泳检测(见图3),约1.2kb处有一条亮带,说明重组质粒上确有扩增的外源基因片段.
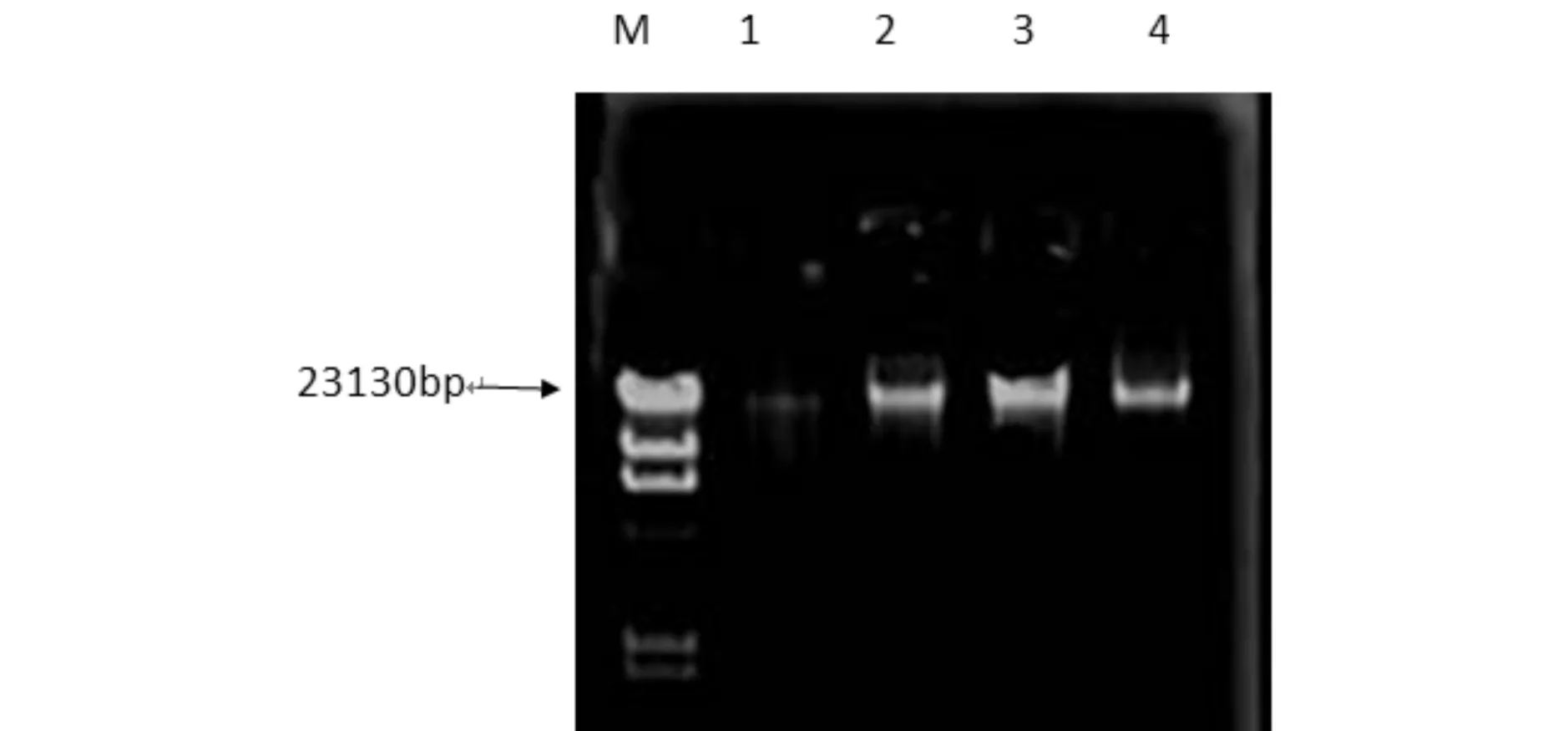
M:λDNA/HindⅢ marker;1-4:H197的总基因组
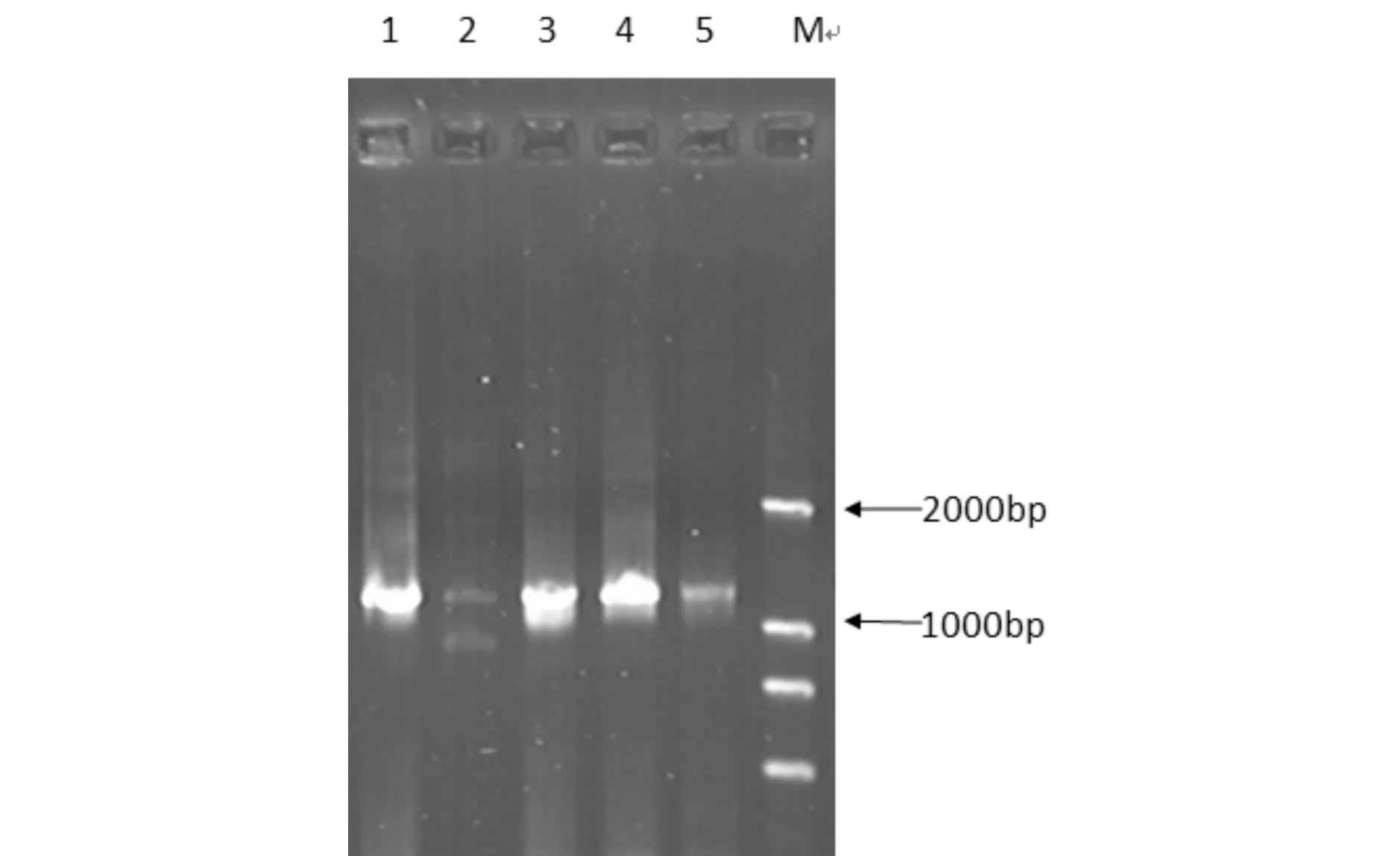
1,3,4,5,6:扩增的目的片段;M:DL2000 marker
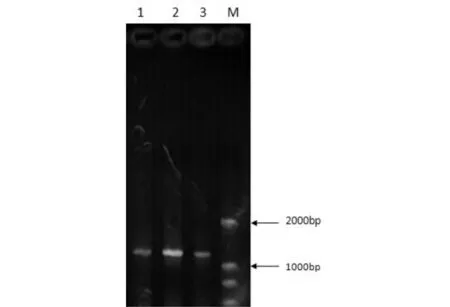
M:DL2000marker;1、2、3:MTG基因
2.2 重组质粒的构建
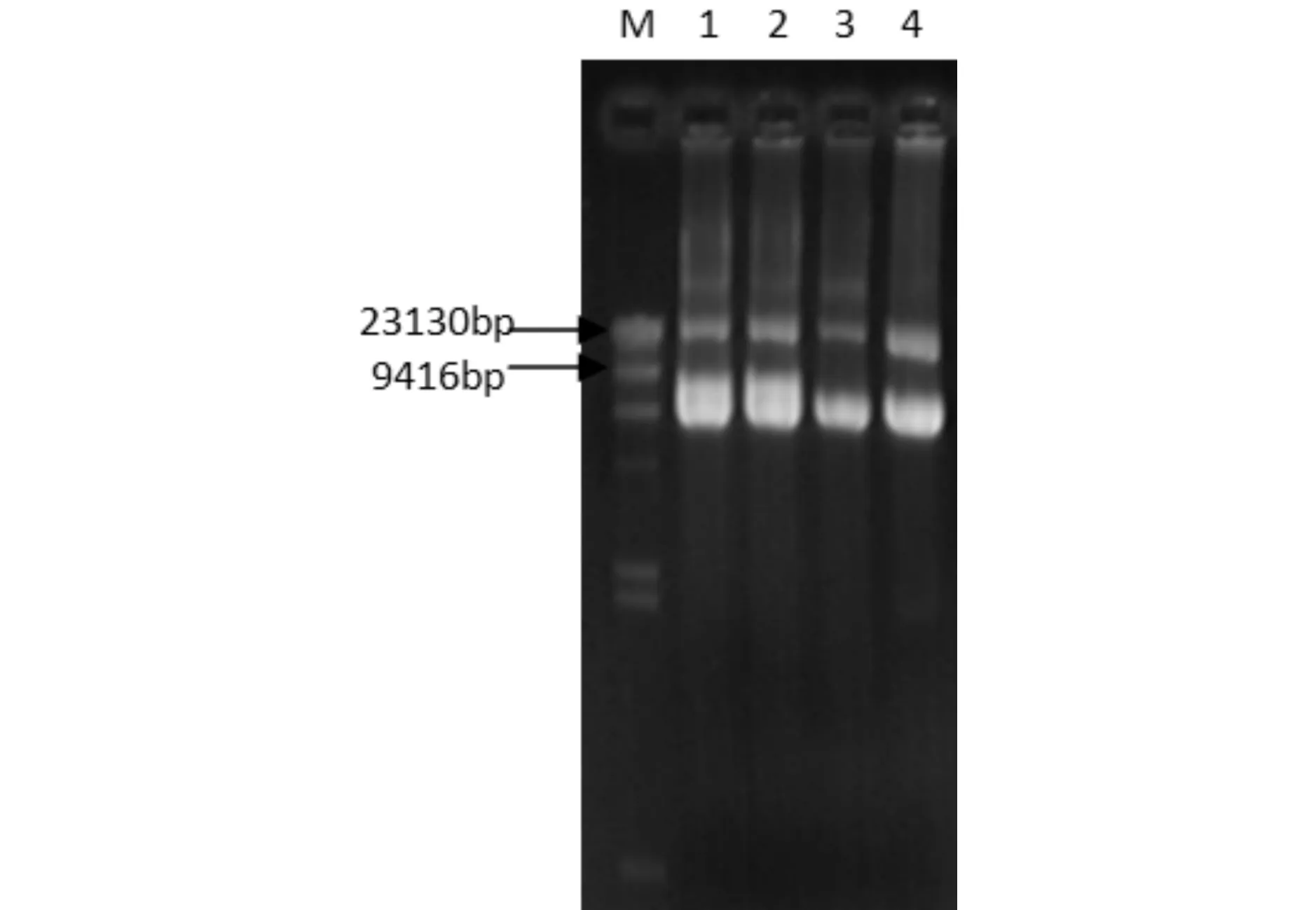
图4为酵母表达载体pPIC3.5k电泳图谱. 由图4可见, 采用碱性裂解法由E.coli提取酵母表达质粒pPIC3.5k,酵母表达质粒处于约9kb附近;图5为载体和目的片段的双酶切电泳图谱. 由图5可见, 利用EcoRI和BamHI分别对PCR产物和pPIC3.5k进行双酶切,酵母表达质粒pPIC3.5k双酶切后位于9kb附近,PCR目的片段位于1.2kb附近,说明扩增和质粒提取均获成功;将酶切后的 MTG基因片段与具有粘性末端的酶切后的pPIC3.5k重组后转入E.coliDH5α细胞中.将转化获得的阳性转化子采用碱裂解法提取质粒并电泳检测(见图6).质粒大小位于10kb附近,说明重组酵母表达质粒pPIC3.5k/MTG构建成功.
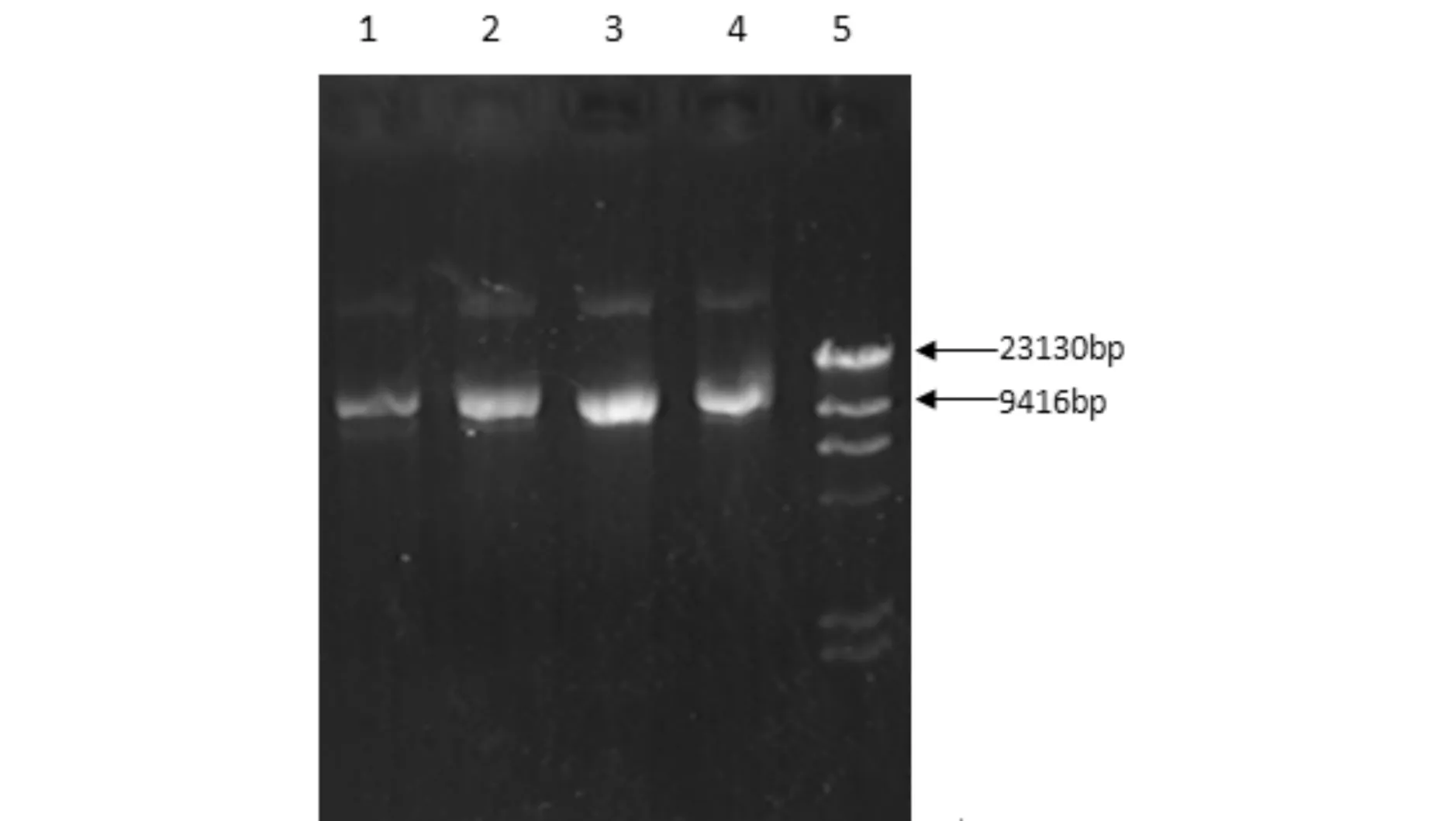
1-4:酵母表达质粒pPIC3.5k;5:λDNA/Hindш Marker
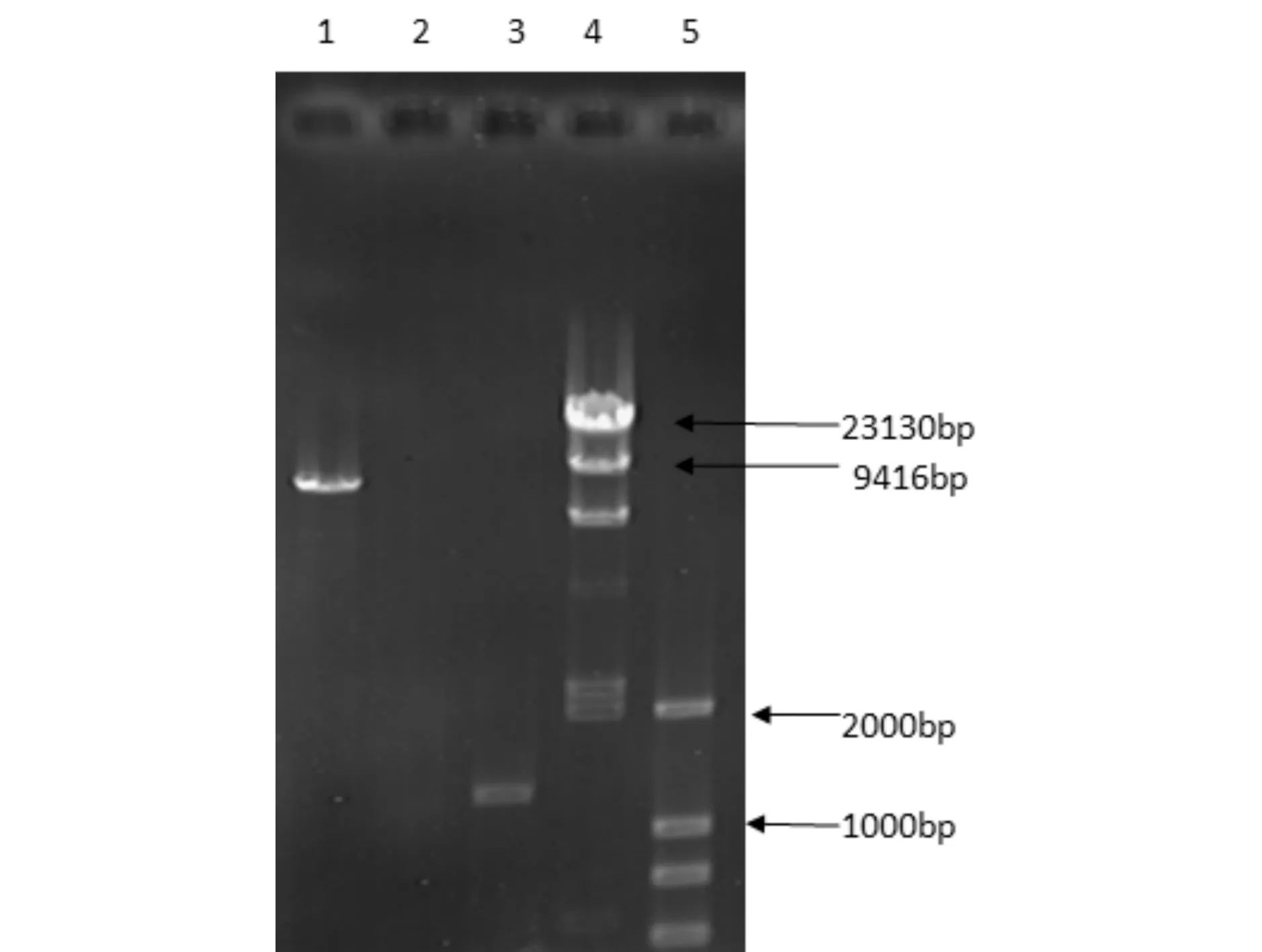
1:pPIC3.5k;2:空泳道;3:目的片段;4:λDNA/ HindⅢ marker;5:DL2000 marker
M:λDNA/HindⅢ maker;1-4:重组质粒pPIC3.5k/MTG
2.3 重组质粒的验证
图7为重组质粒的PCR验证图谱. 由图7可见,将提取的重组质粒进行目的片段的上游引物和下游引物PCR,所得目的片段位于1.2kb附近(见图7中1~4). 对重组质粒鉴定采用上游引物和3'AOX1进行PCR得到位于1.5kb附近的片段(见图7中7~10),证明重组质粒构建成功且方向正确.将提取的重组质粒进行EcoRI和BamHI双酶切,得到位于9kb附近的pPIC3.5k质粒和1.2kb附近的目的片段(见图8),进一步验证重组质粒构建成功.

1-5:上游和下游引物扩增片段;6:DL2000 marker;7-10:上游引物和3'AOX1扩增片段
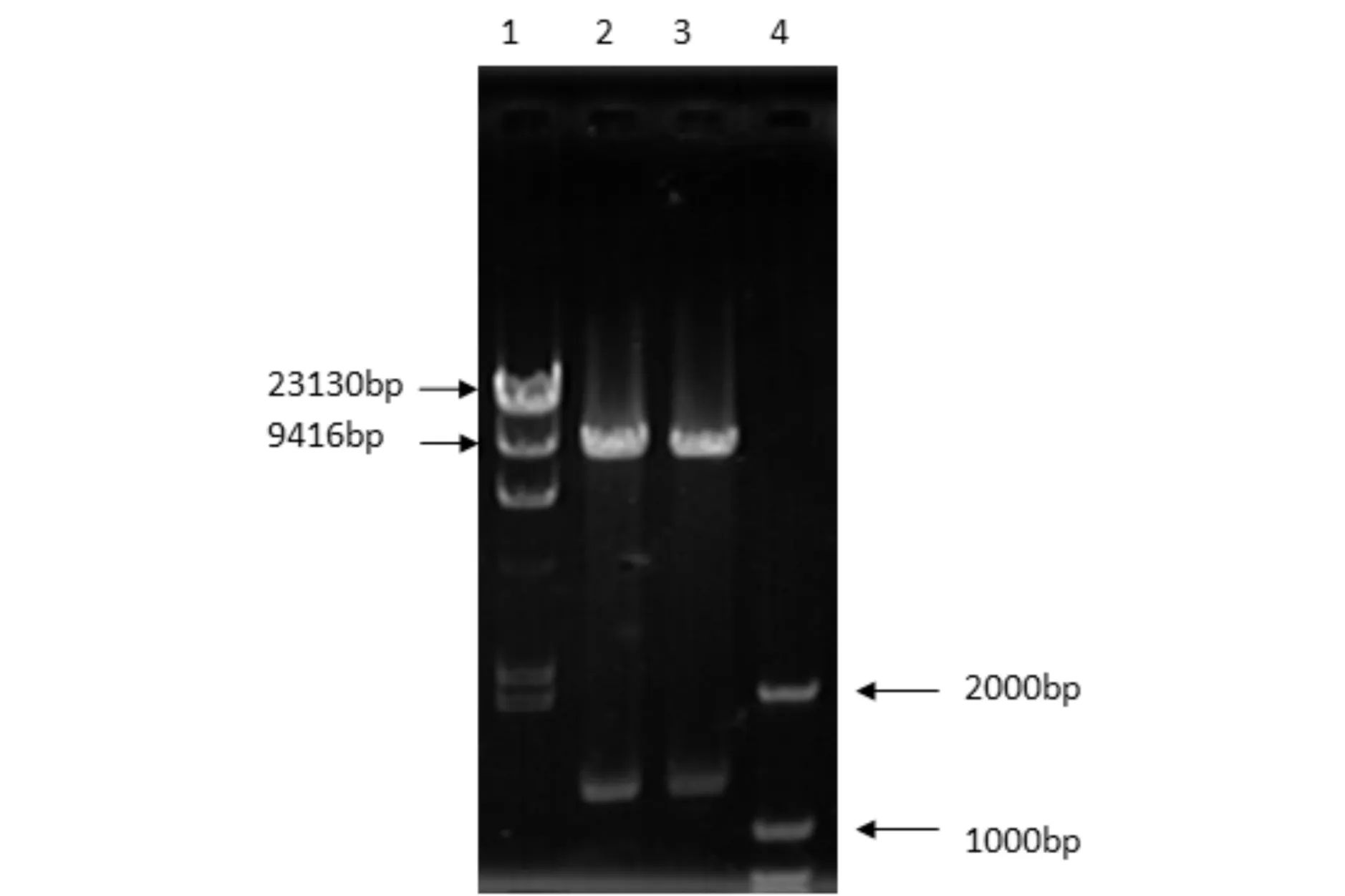
1:λDNA/HindⅢ maker;2-3:重组质粒pPIC3.5k/MTG双酶切;4:DL2000 marker
3 结语
MTG既可催化同种蛋白质之间的交联,也可催化不同蛋白质之间的交联.由于组成各种蛋白质的氨基酸种类互不相同,因而不同的蛋白质中的限制性氨基酸不一定相同,通过MTG可将它们连接起来,实现优势互补,提高蛋白质的功能和营养价值.巴斯德毕赤酵母表达系统是近年来国内外广泛使用的一种新型表达系统[19],该表达系统除具有真核表达系统所具有的特点外,还具有成本低、产量高及利于规模化生产等优点.因此本研究选用pPIC3.5k作为表达载体进行重组质粒构建,将MTG基因插入pPIC3.5k中,通过测序发现,MTG基因已正确连接到pPIC3.5k中,为MTG基因的真核表达奠定了一定的基础.
[1]Folk J E, Finnlayson J. Theε-(γ-glutaminyl)lysine crosslink and the catalytic role of transglutaminases[J].
Adv Prot Chem 1997,31:1-133.
[2]常中义, 江 波.微生物谷氨酰胺转胺酶的应用进展[J].食品科学,2002,21(9):6-8.
[3]Motoki M, Seguro K. Transglutaminase and its use for food processing[J]. Trends Food Sci Technol, 1998,19(9), 204-210.
[4]黄六荣,何冬兰. 微生物谷氨酰胺转胺酶的研究进展[J].微生物学杂志,2003,11(23):52-56.
[5]Ando H, Adadi M, Umeda K et al. Purification and characterisrics of a novel transglutaminase derived from microorganisms[J]. Agricultural Biological Chemistry,1989,53: 2613-2617.
[6]段宇珩.离子束诱变谷氨酰胺转胺酶高产菌株进行选育研究[D]. 郑州: 郑州大学, 2005.
[7]陈国娟,张春红,刘长江.谷氨酰胺转胺酶高产菌株的选育[J].食品科技,2006(2):15-17.
[8]王清路,李俏俏,薛金艳, 等.巴斯德毕赤酵母表达系统的特点及应用[J].生物技术通讯,2006,7(4)640-643.
[9]隋少飞,陈松林.巴氏毕赤酵母表达系统的特点及其研究进展[J].生物技术通报,2004,(3):1-4.
[10]欧阳立明,张惠展,张嗣同.巴斯德毕赤酵母的基因表达系统研究进展[J].生物化学与生物物理进展,2000,27(2):151-154.
[11]常忠义,江 波,王 璋.培养基组成对轮枝链霉菌合成谷氨酰胺转胺酶的影响[J].无锡轻工大学学报, 2001,20(1):51-54.
[12]徐 平,李文均,徐丽华, 等.微波法快速提取放线菌基因组DNA[J]. 微生物学通报,2003,30(4):82-85.
[13]萨姆布鲁克J, 拉塞尔D W. 分子克隆试验指南[M]. 3版. 黄培堂,译. 北京:科学出版社, 2002.
[14]吴乃虎. 基因工程原理[M]. 2版. 北京: 科学出版社, 1998.
[15]吴学敏,单 颖,李 华,等. pGAPZαA/HD5 酵母表达载体的构建[J].中国现代医学杂志,2009, 19(9):2773-2779.
[16]陈 莹, 陈 菁,刘树滔,等.PTD2SOD融合蛋白的毕赤酵母表达载体的构建和表达[J]. 海峡药学,2007,19(7):10-12.
[17]程太平,刘 超,荣 俊.新城疫病毒JZ05 株F 基因重组pGAPZα 的构建[J].湖北农业科学,2009,48(11):2639-2642.
[18]丛 敏,靖学芳,刘振龙,等. pPICZα-synBPIm600酵母表达载体的构建和鉴定[J].首都医科大学学报, 2004,25(1):35-39.
[19]Higgins D R.Overview of protein expression in Pichia pastoris[M].New York:John Wiley and Sons,1995.
猜你喜欢
World Journal of Pediatrics(2023年11期)2023-12-11 01:35:46
食品与发酵工业(2018年3期)2018-04-12 09:35:51
中国调味品(2017年2期)2017-03-20 16:18:25
广东饲料(2016年1期)2016-12-01 03:43:01
工业微生物(2016年5期)2016-11-11 06:58:44
创新作文(小学版)(2016年16期)2016-11-11 05:47:54
化学与生物工程(2016年10期)2016-11-10 06:01:35
现代检验医学杂志(2016年5期)2016-08-20 03:17:04
中外医疗(2015年11期)2016-01-04 03:58:50
中国科技信息(2015年2期)2015-11-16 08:18:32
