
咔唑及其衍生物的电子光谱的理论研究
2011-12-27 03:50刘鸿雁
东北师大学报(自然科学版) 2011年4期
刘鸿雁,李 岩
(吉林化工学院化学与制药工程学院,吉林吉林 132022)
咔唑及其衍生物的电子光谱的理论研究
刘鸿雁,李 岩
(吉林化工学院化学与制药工程学院,吉林吉林 132022)
采用DFT//B3LYP方法,在6-31G基组上对咔唑及其衍生物基态结构进行了优化.计算得到键长、键角、HOMO、LUMO能级.在基态几何优化基础上,用含时密度泛函理论(TD-DFT)计算了吸收光谱,并分析了取代基对咔唑的影响及变化规律.计算结果表明:取代基对键长、键角的影响很小;供电子基使得HOMO和LUMO轨道能量升高,但能隙下降;吸电子基使得咔唑的共轭性程度增强,能隙降低;无论吸电子基团还是推电子基团均使吸收光谱较咔唑发生了红移.
咔唑;衍生物;含时密度泛函;吸收光谱;分子轨道
咔唑及其衍生物分子具有大的共轭体系[1-3],已作为优良的光学材料被深入研究并加以开发应用,如将其作为π给体与π受体形成特殊性能的电荷转移型配合物,将是人们关注的一类新型光电材料.4,9位原子上取代基团无论是给电子基还是吸电子基,直接决定着其衍生物在有机电致发光器件(OLED)中作为空穴传输层或是空穴阻挡层材料.本文研究了咔唑及其衍生物的电子光谱,为其作为光学材料应用提供理论指导.
1 计算模型与方法
运用Gaussian03量子化学程序包,采用DFT方法在B3LYP/6-31G水平上对咔唑及其衍生物的分子结构进行优化,得到了基态稳定构型,其分子模型见图1.

图1 咔唑及其衍生物分子模型
2 结果与讨论
2.1 基态几何构型
用DFT//B3LYP方法,在6-31G基组上优化得到基态构型的一些主要结构参数列于表1和2.由表1和2可见,各取代基对咔唑的键长、键角都有影响,但影响不大.推电子基团使得与取代基相邻C原子与C原子的键长加长.因为推电子基团使得与取代基相连C原子上的电子云密度减少,静电作用减小,所以键长变长.
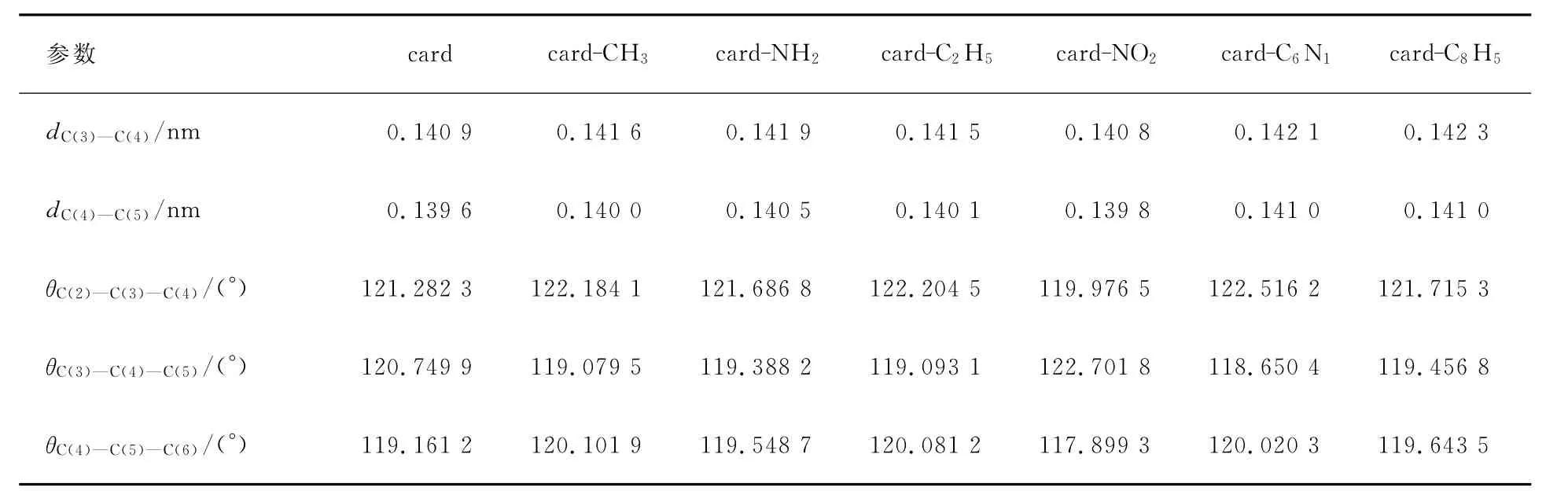
表1 咔唑4位取代基的衍生物的键长、键角
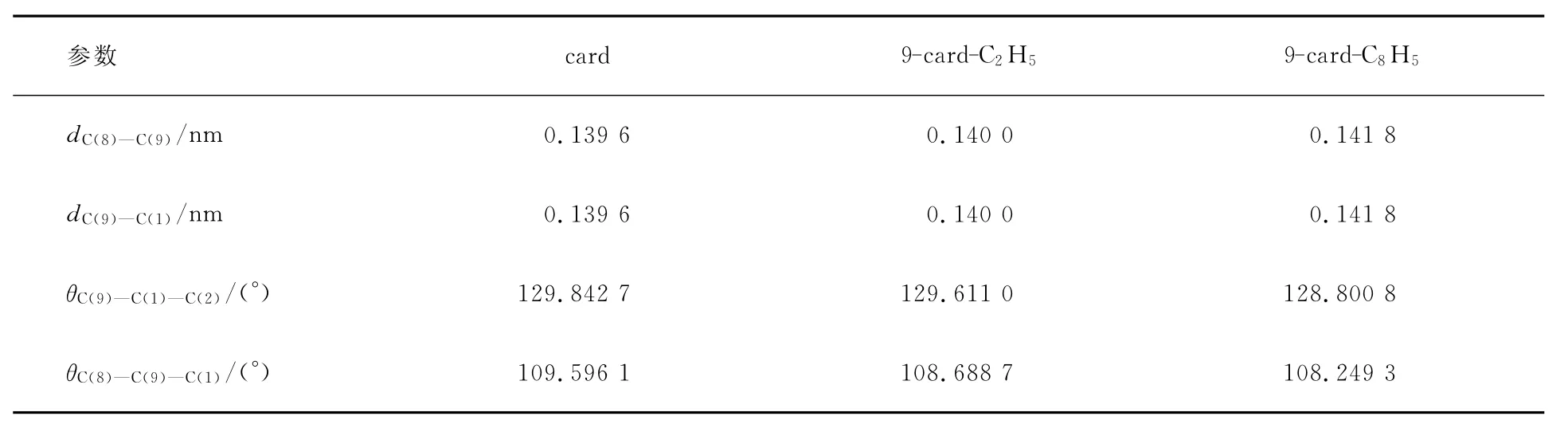
表2 咔唑9位取代基的衍生物的键长、键角
2.2 咔唑及其衍生物的分子轨道分析
发光材料的基态的电子跃迁能量转移对发光效率具有重要的影响[4-6].为了探索咔唑及其衍生物的电子跃迁实质,以我们优化得到的稳定几何构型为基准,对咔唑及其衍生物的分子轨道成分进行了分析,计算得到的前线分子轨道图见图2.
从图2可以看出:在咔唑单体的HOMO轨道上,整个分子的共轭效应较强,主要是苯环上的π轨道与N原子的p轨道形成了p-π共轭,C(11)—C(12)之间存在弱的成键作用.咔唑单体LUMO轨道主要表现反键作用,且N的p电子不再参与共轭,与苯环相连的桥原子C(11)—C(12)的成键作用加强.咔唑衍生物的LUMO轨道也主要表现反键作用,且咔唑环上N的p电子不参与共轭.无论是4位取代还是9位取代,对于推电子基团,在前线轨道间的电子云向咔唑环上转移;而吸电子基团使在前线轨道间的电子云偏离咔唑环,移向吸电子基团,使电子云主要集中在吸电子基团和与之相邻的苯环上.取代基—NO2与—C8H5和—C6N1相比吸电子能力较强,电子云偏移的程度也比较大.对于相同取代基,电子云的转移程度9位取代比4位取代大.
图3为优化得到的各个分子基态的前线分子轨道能级及HOMO与LUMO的能隙.从图3可以看出,咔唑环上的供电子基团使HOMO和LUMO能量升高,HOMO升高的幅度大于LUMO,总的能隙相对咔唑降低.吸电子基团—NO2使HOMO和LUMO能量降低,但LUMO能量降低的程度大于HOMO,总的能隙降低.对于—C6N和—C8H5来说,由于吡啶环和苯环的存在使得HOMO和LUMO能量都升高,HOMO升高的幅度大于LUMO,总的能隙相对咔唑降低.总之,咔唑的衍生物的能隙比咔唑的能隙小,导电能力加强,预示着吸收波长将发生红移.HOMO轨道能量升高,空穴传输能力增强,LUMO轨道能量降低,电子传输能力增强.

图2 咔唑及其衍生物的前线分子轨道

图3 咔唑及其衍生物前线分子轨道能级图
2.3 吸收光谱
优化得到的基态几何构型的电子吸收光谱见表3.其中4,9位咔唑衍生物的主要吸收光谱全部来源于分子中HOMO到LUMO的π-π*的跃迁,最大吸收主要是HOMO-1→LUMO间的跃迁.由表3可知,当咔唑4位被供电子基团—CH3取代后,最大吸收波长红移4.37 nm,被—C2H5和—NH2取代后最大吸收波长分别红移4.61和45.83 nm.由此可以推测出取代基的供电子能力越强,红移程度越大.4位上被—NO2,—C6N1及—C8H5取代后吸收光谱分别红移79.52,53.34和25.74 nm.9位上被—C2H5及—C8H5取代后吸收光谱分别红移10.1和37.45 nm.取代基使咔唑衍生物整个分子的共轭长度增加得越长,红移的范围就越大,含有—NO2强吸电子取代基的咔唑衍生物的最大吸收波长为373.8 nm.总之,4位上的烷基取代对吸收光谱的影响很小.对于相同的取代基,9位比4位上的取代对吸收光谱的影响大.

表3 咔唑及其衍生物的跃迁能、吸收光谱、振子强度及主要跃迁系数
3 结论
无论是4位取代还是9位取代,对于推电子基团取代的分子,在前线轨道间的电子云向咔唑环上转移;而吸电子基团使在前线轨道间的电子云偏离咔唑环移向吸电子基团,使电子云主要集中在吸电子基团和与之相邻的苯环上.取代基—NO2比—C8H5和—C6N吸电子能力强,电子云偏移的程度也比较大.对于相同取代基,9位取代比4位取代电子云的转移程度大.LUMO和HOMO轨道电子云分布不同,这为推测该系列衍生物发光提供了理论基础.4位取代时,对于吸电子基团而言,除—NO2取代外,HOMO轨道能量升高,LUMO轨道能量降低,空穴和电子传输能力均增强.对于推电子基团使前线轨道能量都升高,空穴传输能力增强,电子传输能力减弱.当N原子上的取代基为给电子基团时,有利于提高它的HOMO能级,增加其空穴传输能力.当N原子上的取代基为吸电子基团,则会降低它的HOMO能级及其空穴传输能力.取代基使咔唑衍生物整个分子的共轭长度增加得越长,红移的范围就越大,含有—NO2强吸电子取代基的咔唑衍生物的最大吸收波长为373.8 nm.
[1]DALTONL R,SAPOCHAKL S.Recent advances in non-linear of spectroscopy and nonlinear opticalmaterials[J].J Phys Chem,1993,97:2871.
[2]刘宇芳,陈兆斌,白凤莲.侧链含1,3,4-噁二唑单元和咔唑单元的共聚电致发光材料的合成与表征[J].山西大学学报:自然科学版,2001,24(1):64.
[3]周虹屏,张士润,杨家祥,等.N-乙基-3-咔唑丙烯酸的合成及性质研究[J].化学世界,2003,12:648.
[4]王继芬,封继康,任爱民,等.低聚芴及其衍生物吸收和发射光谱性质的量子化学研究[J].高等学校化学学报,2004,25(4):676-680.
[5]WANG JIFEN,FENG JIKANG,REN AIMIN,etal.Theoretical studies of the absorption and emission properties of the fluorene-based conjugated polymers[J].Macromolecules,2004,37:3451-3458.
[6]LIAO Y,CHEN Y G,SU Z M,etal.TD-DFT study on electronic spectrum property for bepp2 and its related complexes[J].Synthetic Metals,2003,137(1/2/3):1093-1094.
Theoretical studies on electronic spectrum property of carbazole and its derivatives
LIU Hong-yan,LI Yan
(Chemistry and Drugs Manufacture Engineering College,Jilin Institute of Chemistry Technology,Jilin 132002,China)
The ground-state geometries of carbazole and its derivatives were optimized using DFT/B3LYP method at the 6-31G basis set,as implemented in Gaussian 03.And the bond length,bond angle,HOMO,LUMO orbitals and energy levels were obtained.The absorption spectra and the excitation energies were abtained using the time density functional theory(TD-DFT),6-31G base set,at the optimized geometries of the ground states.And the influence of substituting bases to the carbazole are then analyzed.The computed results indicated that the bond length and bond angle are influenced very slightly by substituting bases.Power supply subbasis cause HOMO、LUMO orbital energies to elevate,but make the energy gaps drop relatively.Attracts the electricity subbasis cause the conjugational degree of carbazole to strengthen,but make the energy gaps to drop relatively.Power supply subbasis and attracts the electricity subbasis both make absorption spectra red shift.And absorption spectra are influenced very slightly by alkylations substituting bases on 4 of carbazole.As the same substituting group,the influence to absorption spectra of substituting bases on 9 of carbazole are bigger than substituting bases on 4 of carbazole.
carbazole;derivative;TD-DFT;molecular orbitals
O 641
150·20
A
1000-1832(2011)04-0096-05
2011-06-17
吉林省科技发展计划项目(20111814).
刘鸿雁(1973—),女,硕士,讲师,主要从事物理化学研究.
石绍庆)
猜你喜欢
辽宁化工(2022年8期)2022-08-27
石油炼制与化工(2021年12期)2021-12-14
物理学报(2021年12期)2021-07-01
数学物理学报(2020年6期)2021-01-14
青岛大学学报(工程技术版)(2019年2期)2019-09-10
塑料助剂(2018年6期)2018-03-25
山东化工(2018年1期)2018-03-10
科技资讯(2016年5期)2016-08-13
武汉工程大学学报(2016年1期)2016-04-07
枣庄学院学报(2015年5期)2016-01-09
