
左旋多巴醇在不对称还原中的应用研究
2011-12-21 02:33冯瑞
化工技术与开发 2011年11期
冯 瑞
(河南南阳医学高等专科学校 ,河南 南阳 473058)
左旋多巴醇在不对称还原中的应用研究
冯 瑞
(河南南阳医学高等专科学校 ,河南 南阳 473058)
在 NaBH4/Cd2+、NaBH4/Co2+、NaBH4/Ce3+、NaBH4/Ni3+还原体系中,采用加入手性源左旋多巴醇和不加入左旋多巴醇两种方法,分别对苯乙酮进行不对称还原,结果表明,加入手性源左旋多巴醇后,反应速度快,产率高。同时在这4种还原体系中NaBH4/Ce3+的催化效果最好。
左旋多巴醇;苯乙酮;不对称还原
手性氨基醇化合物在不对称合成中主要用途有2个:手性配体(ligands)和手性助剂(auxiliaries)。手性氨基醇在不对称合成领域是合成手性催化剂的重要手性源[1],在外消旋体拆分等领域也有着广泛的应用[2]。特别是作为手性配体,手性氨基醇中的氧原子及氮原子具有良好的配位能力,它能与过渡金属配位,形成手性催化剂,应用于许多不对称反应中,如不对称还原[3~4],不对称 1,2-加成[5],不对称 1,4-加成[6],不对称烷基化及Reformatsky反应[7]等,可以很好地催化氢化C=O和C=N等键,从而制备手性醇和手性胺[8]。左旋多巴醇是一种新型的手性氨基醇,本文讨论它在不同的过渡金属离子体系中,对前手性物质苯乙酮的不对称还原研究。合成路线如下:
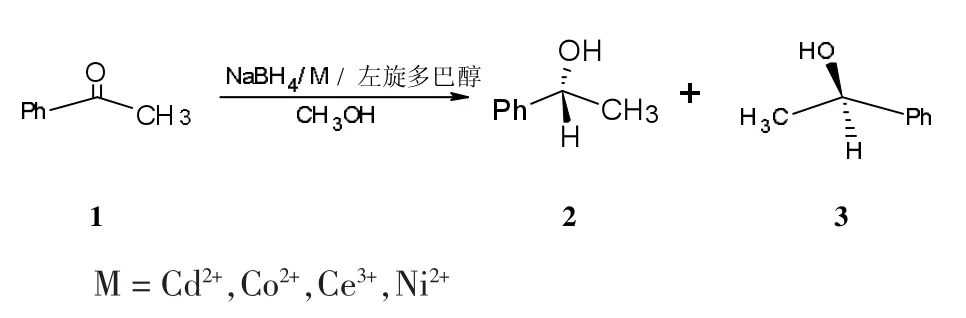
1 实验部分
1.1 主要原料,试剂和仪器
原料苯乙酮和合成中所用其它试剂均为分析纯试剂,溶剂按常规方法进行处理干燥。
R-201型旋转蒸发仪,红外光谱测定采用Nicolet FT-360红外光谱仪,KBr压片。H1核磁共振谱测定采用Vaian 500MHz超导核磁共振仪,CDCl3为溶剂(除特殊注明外),TMS作内标。熔点测定采用X4显微熔点测定仪测定,温度计未经校正,比旋光度采用WZZ-2S数字式自动旋光仪测定。
1.2 合成步骤
1.2.1 加入 CdCl2·5/2H2O
(1)反应体系中加左旋多巴醇时的还原
称取601mg(5mmol)苯乙酮溶于 15mL甲醇中,加入 113mg CdCl2·5/2H2O(0.5mmol),左旋多巴醇 91.5mg(0.5mmol),室温下搅拌约 0.5h,分批加入NaBH4,溶液逐渐变为橙黄色,放出大量气体、放热,TLC跟踪,室温搅拌4h反应结束,用1%HCl淬灭反应,调节 pH=4~5,过滤,用乙醚萃取滤液,合并醚液,饱和食盐水洗涤,用无水Na2SO4干燥。过滤,减压蒸去溶剂,得粗产品。再用硅胶柱层析 (乙酸乙酯∶石油醚=1∶4), 得到未反应原料101mg,得纯物质 508.3mg,为棕红色的油状物质,产率为80%(产率的计算扣除未反应的原料,下同 )。 [α]D30-2.72°(C =1.47mg·mL-1,丙 酮 )。IR(KBr,v/cm-1):3427,2917, 2843, 1446, 1368,1254,1094,1017,800;1H-NMR (500Hz,CDCl3),δ:1.52 (3H,d,J =6.6Hz,2-CH3),4.93 (1H,q,J =6.4,6.5,6.4Hz,1-CH),7.32 (1H,m,J=1.9Hz,p-Ar-H),7.40(4H,dd,J=7.2,7.9Hz,Ar-H)。
(2)没有加左旋多巴醇(合成路线同上)
601mg(5mmol)苯乙酮,113mg(0.5mmol)CdCl2·5/2H2O, 室温下搅拌约 0.5h, 分批加入303mg(8mmol)NaBH4,反应时间 10h,得到360.5mg棕红色的油状物质,化学产率70%。
下面反应同上述(1)的合成方法(即加入左旋多巴醇)相同。
1.2.2 加入 CeCl3·7H2O
加入 601mg(5mmol)苯乙酮,187mg(0.5mmol)CeCl3·7H2O,左旋多巴醇 91.5mg(0.5mmol),室温下搅拌约 0.5h, 分批加入 303mg(8mmol)NaBH4,反应时间2h,分离得到纯产品432mg,化学产率79%,[α]D30-2.76°(C=1.47mg·mL-1,丙酮)。
1.2.3 加入 NiCl2·7H2O
加入 601mg(5mmol)苯乙酮,加入 NiCl2·7H2O 117mg(0.5mmol),左旋多巴醇 91.5mg(0.5mmol),室温下搅拌约 0.5h, 分批加入 303mg(8mmol)NaBH4,反应3h,得纯产品543.8mg,化学产率82%,[α]D30-2.71°(C=1.47mg·mL-1,丙酮)。
1.2.4 加入 CoCl2·6H2O
加入601mg(5mmol)苯乙酮,加入CoCl2·6H2O 180mg (0.5mmol), 左 旋 多 巴 醇 91.5mg(0.5mmol),室温下搅拌约 0.5h,分批加入 303mg(8mmol)NaBH4, 反 应 5h,得 399.4mg,化 学 产 率75%,[α]D30-2.69°(C=1.47mg·mL-1,丙酮)。
2 结果与讨论
2.1 结果与讨论
以CdCl2·5/2H2O为例,在相同的条件下,加左旋多巴醇和不加左旋多巴醇时反应时间和产率的比较,如表1所示。
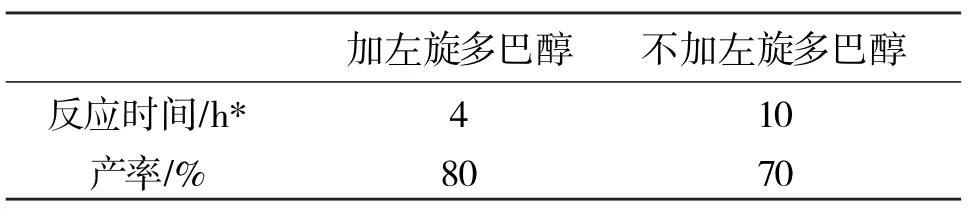
表1 加左旋多巴醇和不加左旋多巴醇对反应的影响
从表1可以看到,加左旋多巴醇比不加左旋多巴醇反应速度明显加快,并且产率也高,可见左旋多巴醇在还原苯乙酮中起到催化作用,大大缩短反应时间。
当原料苯乙酮为 601mg(5mmol),左旋多巴醇 91.5mg(0.5mmol),NaBH4303mg(8mmol)时,在不同过渡金属氯化物催化下对反应的影响见表2。
从表2可以看到,在不同的过渡金属离子氯化物催化下,反应结果有较大的差别,其中以Ce3+为催化剂条件下,还原产物收率最好,反应速率也很快,而以Co2+为催化剂时,收率最差。在测试条件相同的情况下(C=1.47mg·mL-1,丙酮),Ce3+作催化剂时[α]D30最高为-2.76°。
2.2 小结
探讨了在以手性左旋多巴醇及过渡金属离子氯化物催化下对潜手性化合物苯乙酮的不对称还原,研究发现以Ce3+作催化剂时,反应速度最快,而以Co2+为催化剂时,收率最差。原因可能是Ce3+更容易和左旋多巴醇形成配位化合物,形成手性催化剂,可以很好地催化氢化前手性物质苯乙酮C=O键,从而制备手性醇1-苯基乙醇。
[1] Corey E J , Helal C J.Reduxtion of carbonyl compounds with chiral oxazaborolidine catalysts:A new paradigm for enantioselective catalystsis and a powerful new synthetic method [J].Angew .Chem .Int.Ed.,1998 ,37 (15):1986-2012.
[2] Kawai M,Omori Y,Yamamuru H,et al.Optical resolution of N-carbenzoxy-a-methoxyglicine[J].Tetrahedron:Asmmestry, 1992, 3(8):1019-1020.
[3] Shinichi Itsuno, Koichi Ito,A Lira Hirao ,et al.A symmetric reduction of aliphatic ketones with the reagent prepared from (s)-(-)-2-Amino-3-methy-1,1-diphenybutan-1-ol and borane[J].Org Chem,1984,49:555-557.
[4] Itsuno S, Hirao A,Nakaham S, et al.A symmetric synthesis using chairally modified borohydridePart 1 Euantio selective reduction of aromatic ketones with the reagent prepared from borane and (s)-valinol [J]J.Chem Soc,Perkin Trans1,1983:1673-1667.
[5] Jan G Batelaan, Hendrk JHageman, Jan Verbeek.The photodecomposition of ocnitratomethyldenzoin as studied by ESR[J].Tetrahedron Lett,1987,28:2163-2166.
[6] Kenso Soai,Yasuhiro Kawase.A symmetric synthesis of β-hydroxyesters by the enantio selective refomatsky reaction in the presence of chiral aminoalcohols[J].Te-trahedron A symmetry,1991,(2):781-784.
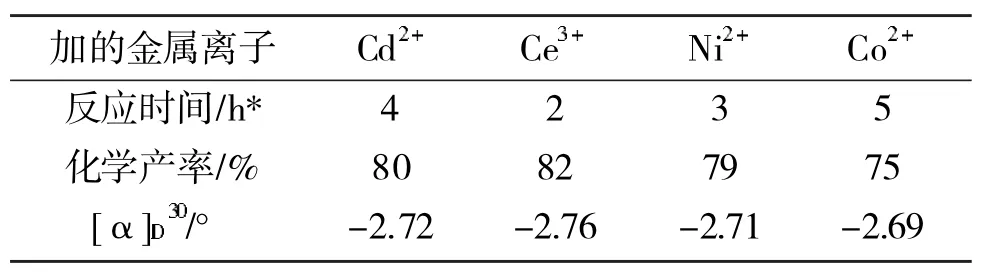
表2 在不同过渡金属氯化物催化下对反应的影响
[7] Kenso Soai, Yuji Hirose;Shuichi Sakata. Highly enautio selective synthesis of β-hydroxy nitriles by the cyanocn ethylation of aldehydes using DPM as achiral.
[8] 刘二畅,杨定娇,董建霞.现代手性药物的合成与发展[J].广东药学院学报,2004,20(6):685-688.
Applied Research on L-dopa Alcohol in Asymmetric Reduction
FENGRui
(Nanyang Medical College, Nanyang473058,China)
Asymmetric reduction of acetophentone was prepared by two methods of adding L-dopa Alcohol or no adding L-dopa alcohol with NaBH4/Cd2+、NaBH4/Co2+、NaBH4/Ce3+、NaBH4/Ni3+as the reducing agent.The optimum conditions were as followed:the method of adding L-dopa alcohol had the advantages of faster reaction speed and higher yield.At the same time, with NaBH4/Ce3+as the reducing agent was the best of catalytic effect in the four reduction system.
L-dopa alcohol;acetophentone;asymmetric reduction
T Q 031
A
1671-9905(2011)11-0005-02
广西科学基金项目资助(桂科基0575055)
冯瑞(1975-),女,讲师,硕士,主要从事有机合成的研究,电话:13525107689,E-mail:fengrui20032006@163.com
2011-08-08
猜你喜欢
云南化工(2021年11期)2022-01-12
广东医科大学学报(2020年4期)2020-08-24
理化检验-化学分册(2020年2期)2020-06-01
消费导刊(2019年14期)2019-08-21
消费导刊(2019年27期)2019-07-22
表面工程与再制造(2019年1期)2019-05-11
合成化学(2015年2期)2016-01-17
中国当代医药(2015年30期)2015-03-01
物理化学学报(2015年5期)2015-02-28
中国药业(2014年19期)2014-05-17
