
新型可溶性酞菁的合成和光致发光及电致发光性质
2011-11-30 10:41:40白青龙张春花程传辉李万程申人升杜国同
物理化学学报 2011年5期
白青龙 张春花 程传辉 李万程 申人升 杜国同,,*
(1大连理工大学物理与光电工程学院,辽宁大连116024;2内蒙古民族大学化学化工学院,内蒙古通辽028043; 3吉林大学集成光电子国家重点实验室,电子科学与工程学院,长春130012)
新型可溶性酞菁的合成和光致发光及电致发光性质
白青龙1,2张春花2程传辉1,*李万程3申人升1杜国同1,3,*
(1大连理工大学物理与光电工程学院,辽宁大连116024;2内蒙古民族大学化学化工学院,内蒙古通辽028043;3吉林大学集成光电子国家重点实验室,电子科学与工程学院,长春130012)
以3(4)-硝基邻苯二腈和对甲氧基苯酚为原料,经过两步反应合成了α(β)-四(4-甲氧基苯氧基)酞菁锌,通过谱学方法和元素分析表征了其结构.比较研究其溶液和旋涂膜的紫外可见光谱、光致发光光谱和固体薄片的光致发光光谱.并以其旋涂膜为发光层制备了电致发光器件,研究其电致发光性质.结果表明,固态酞菁材料与其溶液的荧光发射波长相比均向长波方向移动了145 nm以上,而且都有不同程度的宽展.在固态下β-位取代酞菁荧光发射波长红移的程度比α-位取代酞菁大.电致发光光谱的发射波长和其旋涂膜的光致发光光谱的发射波长基本一致,大约在856和862 nm左右.在固态下酞菁分子排列紧密,分子间相互作用增强导致了荧光发射波长的巨大红移.
可溶性酞菁;合成;光致发光;电致发光;红移
1 引言
酞菁化合物是一类非常重要的有机染料,从1907年Braun和Tcherniac1合成了第一个非金属的酞菁化合物以来,经过几十年的发展,酞菁已发展成为一门独立的学科.酞菁和酞菁配体具有特殊的二维共轭π电子结构,共轭的大环体系有强烈的π-π电子作用,这是该类化合物具有特殊的光、电、磁学等特殊性质的理论基础.在非线性光学材料、2光限制配合物材料、3,4分子电子元件、5,6电致变色显示材料、7,8液晶显示材料、9,10光动力学癌症治疗药物11-13等一些列领域具有巨大的潜在的应用价值.
而非取代酞菁几乎不溶于常见的有机溶剂,不便于研究其光学性质和电学性质等,而且不能用旋涂的方法成膜,只能用真空蒸镀的方法成膜.14但是,可以通过在酞菁外围苯环上引入一些取代基,如烷基、芳基或杂环基等15,16的方法提高其溶解性能.其中取代基的类型、大小、数目和位置等都能够很容易地通过化学方法来进行修饰和调整,而且通过调整外围取代基的结构和组成还可以实现对酞菁材料光学性质和电学性质的调制.
我们在酞菁锌外围苯环的α和β-位上引入了甲氧基苯氧基取代基合成了两种易溶于常见有机溶剂的酞菁锌衍生物α和β-四(4-甲氧基苯氧基)酞菁锌(简称α和β-TMPPcZn),研究其溶液和旋涂膜的吸收光谱和光致发光光谱,以及固体薄片的光致发光光谱.并将酞菁材料作为发光客体按一定的比例掺入主体发光材料聚乙烯咔唑(PVK)中,以其旋涂薄膜作为发光层,制备了电致发光器件,研究其电致发光性质.
2 实验部分
2.1 仪器与试剂
UV-2450型紫外-可见(UV-Vis)分光光度计(日本);Nicolet-20DXB型红外(IR)分光光度计(美国); Bruker Arance II400M型核磁共振(NMR)仪(瑞士); varioEL III元素分析仪(德国);ASIMA CFR型飞行时间质谱(MS(LDI-TOF))仪(英国);DZF-6020型真空干燥箱(上海一恒科技);KW-4A型台式匀胶机(中国科学院);SO400型-多源自动控温有机分子气相沉积系统(沈阳四达);DM-300B型镀膜机(北京北仪);Keithly 2400 Source Meter(美国).
3-硝基邻苯二腈和4-硝基邻苯二腈(分析纯,日本,东京化成工业株式会社);1,8-二氮杂双环[5,4, 0]-7-十一烯(DBU)(分析纯,日本,东京化成工业株式会社),其它试剂均为分析纯(天津科密欧化学试剂有限公司).
2.2 合 成
2.2.1 3-(4-甲氧基苯氧基)邻苯二腈的合成
将对甲氧基苯酚(1.24 g,10 mmol)和3-硝基邻苯二腈(1.73 g,10 mmol)溶解在20 mL干燥的二甲基亚砜(DMSO)中,边搅拌边加入氢氧化锂(0.42 g, 10 mmol),氮气保护下,在40°C搅拌48 h,反应混合物冷却后倒入200 mL冰水中析出沉淀,抽滤,滤饼用去离子水洗涤三次,再用少量的甲醇洗涤一次,自然风干,溶于少量丙酮中,柱层析分离提纯(流动相为乙醚:石油醚=3:1(体积比))得淡黄色晶体,1.71 g,产率68.4%.
IR(KBr),ν:2230 cm-1(s,CN),1246 cm-1(vs,C―O―C);UV-Vis(CHCl3),λmax:319.5 nm;1H NMR (C3D6O,500 MHz),δ:7.797-7.838(t,J1=8.8.457,J2= 7.955,1H,ArH),7.689-7.708(d,J=7.574,1H,ArH), 7.184-7.217(d,J=9.013,3H,ArH,Ar2H),7.054-7.077(d,J=9.054,2H,Ar2H),3.845(s,3H,CH3). C15H10N2O2元素分析计算结果(w,%,下同):C,71.99, H,4.03,N,11.19;实验结果(%):C,69.68;H,3.12; N,13.07.
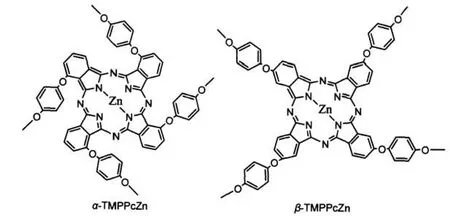
图1 α-和β-TMPPcZn的分子结构Fig.1 Molecular structures of α-and β-TMPPcZn
2.2.2 4-(4-甲氧基苯氧基)邻苯二腈的合成
将对甲氧基苯酚(1.24 g,10 mmol)和4-硝基邻苯二腈(1.73 g,10 mmol)溶解在20 mL干燥的DMSO中,边搅拌边加入氢氧化锂(0.42 g,10 mmol),在氮气保护下,35°C下搅拌48 h.反应混合物冷却后倒入200 mL冰水中析出沉淀,抽滤,滤饼用去离子水洗涤三次,再用少量甲醇洗涤一次,自然风干,溶于少量丙酮中,用柱层析分离提纯(流动相为乙醚:石油醚=7:4(体积比)),得淡黄色晶体, 1.79 g,产率71.6%.
IR(KBr),ν:2231 cm-1(s,CN),1247 cm-1(vs,C―O―C);UV-Vis(CHCl3),λmax:305.5 nm;1H NMR (C3D6O,500 MHz),δ:7.986-8.008(d,J=8.780,1H, ArH),7.532-7.538(d,J=2.566,1H,ArH),7.355-7.384(dd,J1=2.372,J2=6.197,J3=3.567,1H,ArH), 7.148-7.171(d,J=9.110,2H,ArH),7.046-7.069(d,J= 9.096,2H,ArH),3.842(s,3H,CH3).C15H10N2O2元素分析计算结果(%):C,71.99;H,4.03;N,11.19;实验结果(%):C,69.71;H,3.03;N,13.12.
2.2.3 α-(4-甲氧基苯氧基)酞菁锌的合成
将3-(4-甲氧基苯氧基)邻苯二腈(1.00 g,4 mmol)和乙酸锌(0.22 g,1 mmol)加入10 mL干燥的正戊醇中,边搅拌边加入催化剂量的DBU,在氮气保护下,迅速升温至135℃加热搅拌8 h,冷却到室温,在反应混合液中加入无水甲醇析出绿色絮状沉淀,减压抽滤除出有机溶剂,滤饼用甲醇洗涤三次,所得固体自然风干后放入索氏提取器,分别用无水甲醇和去离子水抽提48 h至提取液澄清透明,再用柱层析分离提纯(流动相为乙酸乙酯:乙醚=7:5(体积比))得草绿色粉末0.459 g,产率43%.
IR(KBr),ν:1609 cm-1(m,C=N),1238 cm-1(s, C―O―C),748(s,Pc),879 cm-1(w,Zn-N);UVVis(CHCl3),λmax:329,365,626,694 nm;1H NMR (CDCl3,500 MHz),δ:9.174-9.074(m,1H,ArH),8.813-8.690(m,1H,ArH),8.154-7.955(m,2H,ArH),7.710-7.630(m,4H,ArH),7.480-7.378(m,4H,ArH),7.229-6.886(m,16H,ArH),3.815-3.751(m,12H,CH3); MS(LDI-TOF)m/z:1066.93.C60H40N8O8Zn元素分析计算结果(%):C,67.58;H,3.78;N,10.51;实验结果(%):C,67.49;H,3.62;N,10.68.
2.2.4 β-(对甲氧基苯氧基)酞菁锌的合成
将4-(4-甲氧基苯氧基)邻苯二腈(1.00 g,4 mmol)和乙酸锌(0.22 g,1 mmol)加入10 mL干燥的正戊醇中,边搅拌边加入催化剂量的DBU,在氮气保护下,迅速升温至120°C加热搅拌8 h,冷却到室温,在反应混合物中加入无水甲醇析出绿色絮状沉淀,减压抽滤除去有机溶剂,滤饼用甲醇洗涤三次,所得固体自然风干后放入索氏提取器,分别用无水甲醇和去离子水抽提48 h至提取液澄清透明,再用柱层析分离提纯(流动相为乙酸乙酯:乙醚=7:3(体积比)),得绿色粉末0.479 g,产率45%.
IR(KBr),ν:1605 cm-1(m,C=N),1222 cm-1(s, C―O―C),746 cm-1(s,Pc),889 cm-1(w,Zn―N); UV-Vis(CHCl3),λmax:358,611,679 nm;1H NMR (CDCl3,500 MHz),δ:8.614-8.734(m,4H,ArH), 8.216(s,2H,ArH),8.271(s,2H,ArH),7.446-7.619 (m,12H,Ar4H,Ar8H),7.168-7.260(m,8H,ArH), 3.896-3.924(m,12H,CH3);MS(LDI-TOF)m/z: 1067.80(M+H)+.C60H40N8O8Zn元素分析计算结果(%):C,67.58;H,3.78;N,10.51;实验结果(%):C, 67.43;H,3.61;N,10.68.1
2.3 酞菁发光材料的制备和测试
2.3.1 旋涂膜的制备
将酞菁材料和PVK以3:7(质量比)比例混合,溶于三氯甲烷中,其中酞菁材料的浓度为1 g·mL-1, PVK的浓度为2.3 g·mL-1.以3000 r·min-1的速率将其旋涂在1 cm×1 cm的石英玻璃片上,将涂好的片子放置在真空干燥箱中保存,备用.
2.3.2 酞菁固体薄片的制备
分别将酞菁材料与溴化钾烘干,按1:50(质量比)的比例混合研细后压成直径10 mm,厚度约为2 mm的薄片,在真空干燥箱中保存,备用.
2.3.3 光致发光性质的测试
将样品用专用胶粘在样品室的靶上,以激发波长为532 nm的Nd-YAG激光器作为光源,由液氮冷却(77 K)的锗探测器接受信号,经锁相放大器后由电脑记录和处理实验数据.所有测试均在大气中进行.
2.4 电致发光器件的制备和测试
2.4.1 电致发光器件的制备
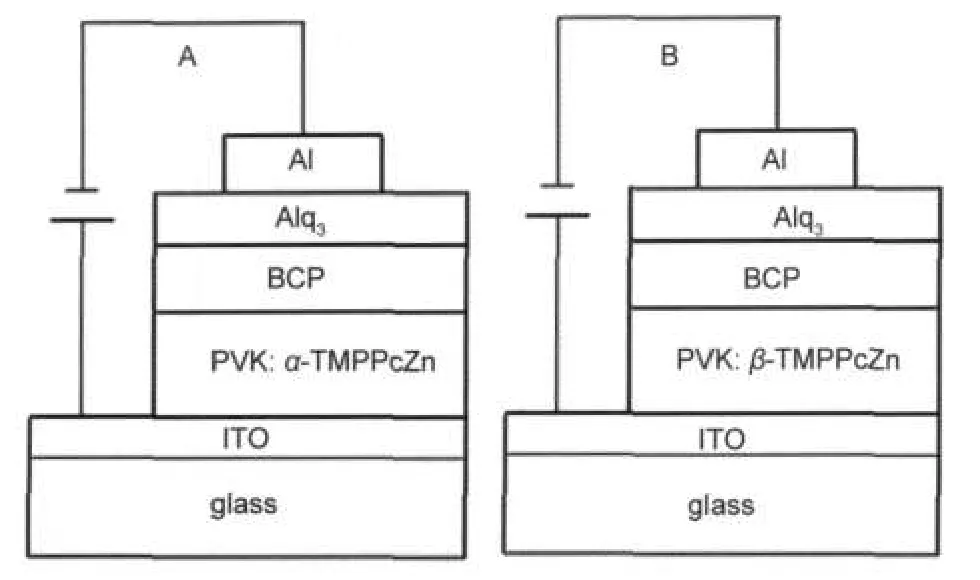
图2 电致发光器件A和B的结构Fig.2 Structures of electroluminescent devices ofAand B

图3 电致发光器件A和B的电流密度-电压特性Fig.3 Current density-voltage characteristics of electroluminescent devices ofAand B
将ITO玻璃衬底(约20 Ω·□-1)分别用丙酮和乙醇棉球反复擦洗干净,然后分别在丙酮、乙醇和去离子水中各超声10 min充分清洁,吹干.将酞菁材料与PVK按3:7的比例混合,将它们溶于三氯甲烷中配制成一定浓度的溶液,然后以3000 r·min-1的速率将它在ITO玻璃的表面上旋涂40 nm厚的薄膜,其厚度由椭偏仪测量.将其放入真空干燥箱中在50°C处理1 d.然后在它上面蒸镀一层20 nm厚的2,9-二甲基-1,10-邻菲罗啉(BCP)为空穴阻挡层,接着生长一层15 nm厚的8-羟基喹啉铝(Alq3)薄层来传输和注入电子.再蒸镀一层120 nm的金属铝作为阴极.器件结构如图2所示.
图3为α和β-TMPPcZn掺杂PVK器件的电流密度-电压特性.实验中器件在这些驱动电压下能够稳定工作.
2.4.2 电致发光性质的测试
在器件的ITO和Al电极上加电压,由液氮(77 K)冷却的锗探测器接受信号,经锁相放大器后由电脑记录和处理实验数据.测试时锗探测器前放置800 nm的滤光片滤除PVK和Alq3等发出的光.整个实验过程在暗室中进行.
3 结果与讨论
3.1 酞菁材料的结构和溶解性
四取代酞菁实际上是由四种同分异构体组成的混合物,异构体的分子对称性分别是D2h,Cs,C2v, C4h.根据文献报道,17,18四取代酞菁的同分异构体有一个理论上的存在比例,即D2h:Cs:C2v:C4h=1:4:2:1(摩尔比),这个理论上的存在比例受中心金属反应条件等因素的影响会发生变化.有人用高压液相成功分离过几种四取代酞菁的同分异构体.19,20本文是用普通的柱层析法分离提纯的,所得到的结论是基于TMPPcZn四种同分异构体共同存在的基础上的.
TMPPcZn外围苯环上的四个甲氧基苯氧基有效地提高了它在有机溶剂中的溶解性,在氯仿、二氯甲烷、四氢呋喃(THF)、二甲基甲酰胺(DMF)、二甲基亚砜(DMSO)和甲苯等溶剂中都有良好的溶解性,这主要取决于取代基在有机溶剂中的溶解性.此外,TMPPcZn分子外围取代基的空间位阻效应有效地阻止了酞菁分子间的聚集行为,也有利于提高其在有机溶剂中的溶解性.
3.2 酞菁材料的紫外-可见光谱
3.2.1 酞菁溶液的紫外-可见光谱
α-和β-TMPPcZn在DMF中的紫外可见光谱见图4,甲氧基苯氧基取代基的引入对Q带最大吸收波长的影响较大,分别出现在693和679 nm处,与无取代酞菁锌(669 nm)相比都有较大的红移.其中α-TMPPcZn红移程度比β-TMPPcZn大约14 nm,因为HOMO的α-位置的原子轨道线形组合(LCAO)系数比β-位置的大,所以在α-位上引入的取代基对HOMO的作用比β-位的更强,结果导致了HOMOLOMO之间的能级差减小,Q带最大波长红移程度变大.17-19

图4 α-和β-TMPPcZn溶液及其旋涂膜的紫外-可见光谱Fig.4 UV-Vis spectra of α-and β-TPPcZn in DMF and spin-coated films
这两个特征吸收带的出现表明酞菁环的形成,而出现在626和611 nm处的小峰不是由二聚体引起的,是两个酞菁的电子振动峰.21α-和β-TMPPcZn的紫外可见光谱表明了它们在稀溶液(8.85×10-5mol·L-1)中不易聚合的性质,主要原因是其分子外围大体积的甲氧基苯氧基取代基的空间位阻有效地增大了酞菁分子π共轭体系间的距离,使其不易通过π-π吸引而发生分子聚合现象.
3.2.2 酞菁旋涂膜的紫外-可见光谱
由图4可知,酞菁旋涂膜的紫外可见吸收光谱与其溶液的紫外可见光谱很相似,显示了酞菁典型的B带和Q带.两个酞菁的旋涂膜吸收谱Q带的位置与其溶液吸收谱的Q带的位置基本一致,约在696和679 nm处,其主要的区别是和溶液中的吸收峰相比旋涂膜的吸收峰变宽一些.酞菁体系中的聚集可以通过光学吸收谱里最大消光系数的降低和Q带的蓝移来判断.15旋涂膜Q带的位置与DMF溶液Q带的位置变化较小,没有明显的蓝移现象.以上两点足以说明这两种酞菁分子在旋涂膜中虽然有一定的聚集,但其聚集性很弱.这可能是较大的外围取代基团的空间位阻效应阻碍了酞菁分子之间的π-π共轭体系间的相互作用.16而根据分子激子理论中的Davydov规则,22聚集体中从基态到激发态允许的跃迁数等于聚集体单元所含分子数.由于旋涂膜聚集能力很弱,所以其吸收谱宽展的程度也不大.
3.3 酞菁材料的光致发光光谱
3.3.1 酞菁溶液的荧光光谱
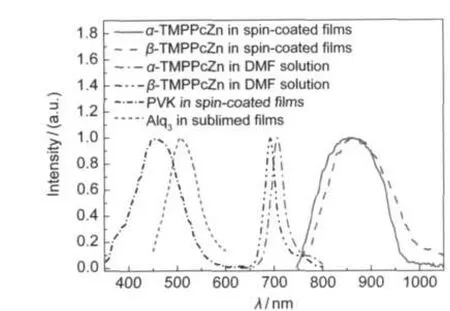
图5 α-和β-TMPPcZn薄膜、Alq3薄膜和PVK薄膜的光致发光光谱Fig.5 Photoluminescence spectra of α-and β-TMPPcZn in film,Alq3in film and PVK in film
α-和β-TMPPcZn在DMF中的荧光光谱(8.85× 10-5mol·L-1,激发波长为610 nm),如图5所示.两者的形状相似,与相应的吸收光谱呈镜像关系,并出现Stokes位移.荧光辐射跃迁都是从S1→S0,由于振动驰豫和内转换等过程需要耗费电子所吸收的光子能量,荧光辐射的能量小于吸收的光子能量,所以,酞菁荧光光谱的峰值相对于其电子吸收光谱红移.其中α-TMPPcZn荧光发射峰在710 nm处, β-TMPPcZn荧光发射峰在692 nm处.说明取代基引入的位置对酞菁荧光光谱性质的影响非常显著.根据分子轨道理论计算结果表明,23取代基在α-位时的HOMO(A1u)轨道的LCAO系数比取代基在β-时的LCAO系数大,因此取代基在α-位时比取代基在β-位时的HOMO贡献更大,进而降低了激发单重态的能级,使得其最大发射波长红移,因此α-位取代酞菁比相应的β-位取代酞菁的Stokes位移程度大.
3.3.2 酞菁旋涂膜的光致发光光谱
由图5可知,两种酞菁旋涂膜的光致发光光谱分别在855和859 nm处有较宽的荧光发射峰,与相应的两种酞菁溶液的荧光发射峰相比,分别向长波方向移动了约145和167 nm.这是由于在固态下酞菁分子的存在状态不同于溶液,固态下酞菁分子之间的有效距离很近,分子之间的相互作用很强,导致了酞菁分子轨道HOMO-LOMO之间的能带变窄,其荧光发射波长发生巨大的红移.另外,酞菁旋涂膜的光致发光光谱与其吸收光谱不同,β-TMPPc-Zn的发射波长红移程度比α-TMPPcZn的大.这与酞菁的分子构型有关.对于金属酞菁来说,其分子构型取决于中心金属离子的大小和酞菁配体的空间位置.对于α-和β-TMPPcZn来说,中心离子半径很小,所以分子构型主要是由取代基的空间位置决定.当取代基在β-位时,空间位阻较小,酞菁分子共轭平面扭曲变形的程度较小,分子之间有效距离近,相互作用强,所以其荧光发射波长红移程度变大.与此相反,取代基在α-位时空间位阻较大,酞菁分子平面性差,分子间有效距离变大,分子间相互作用减弱,所以其荧光发射波长红移的程度比β-TMPPcZn小.
3.3.3 酞菁固体薄片的光致发光光谱
酞菁固体薄片的光致发光光谱见图6.α和β-TMPPcZn的最大发射波长分别红移到884和918 nm处.其最大发射波长红移的程度比旋涂膜还要大.这主要是因为在固体薄片中,酞菁分子容易形成聚集体,聚集体中的分子间距离更近,相互作用更强,其结果是荧光发射波长红移程度更大,所以其荧光发射峰是属于聚集体的荧光峰.与其旋涂膜类似,β-TMPPcZn的荧光发射波长红移的程度比α-TMPPcZn的大.
3.4 酞菁薄膜器件的电致发光光谱
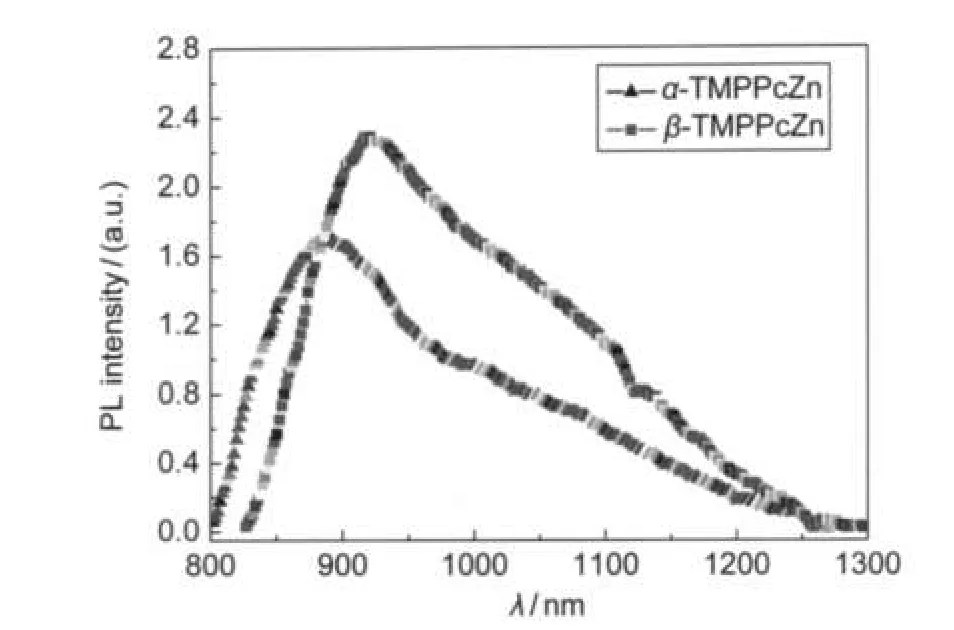
图6 α-和β-TMPPcZn酞菁薄片的光致发光光谱Fig.6 Photoluminescence spectra of α-and β-TMPPcZn in thin disk solid state
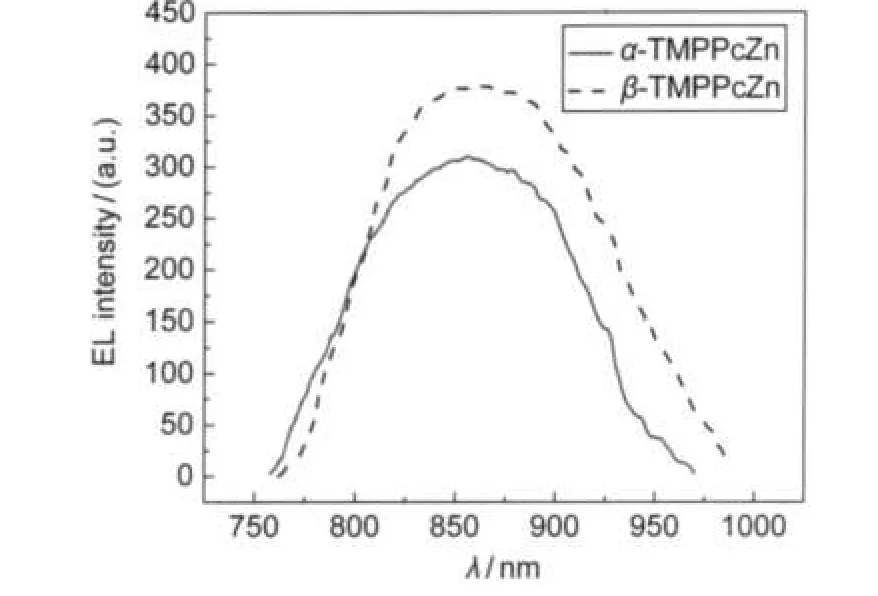
图7 器件A和器件B的电致发光光谱Fig.7 Electroluminescence spectra of devicesAand B
制备了如下结构的两个器件:ITO/PVK: α-TMPPcZn/BCP/Alq3/Al(器 件 A)和 ITO/PVK: β-TMPPcZn/BCP/Alq3/Al(器件B).图7是在10 mA工作电流时PVK中掺杂α-和β-TMPPcZn器件的电致发光光谱.在图中可以看到两种酞菁的发射峰分别在856和862 nm附近,波长范围与其旋涂膜的光致发光光谱基本一致.其中器件B的最大发射波长红移程度比器件A的大,而且器件B的发射峰的强度也比器件A的强.这一点与其旋涂膜的光致发光光谱类似,也是由于作为发光层的两种酞菁材料的分子平面性不同而造成的.
4 结论
α-和β-四(4-甲氧基苯氧基)酞菁锌分子在旋涂膜中虽然有一定程度的聚集,但其聚集性很弱.在旋涂膜中由于分子间的相互作用较强,其光致发光光谱在855和859 nm处有较宽的荧光发射峰,与相应的酞菁溶液相比,分别向长波方向漂移了145和167 nm.固体薄片中酞菁分子主要以聚集体的形式存在,分子间的相互作用更强,其发光峰分别出现在884和918 nm处,与相应的酞菁溶液相比,分别向长波方向漂移了174和226 nm.在固态下β-位取代酞菁荧光发射波长红移的程度比α-位取代酞菁大.
电致发光光谱的发射波长和其旋涂膜的光致发光光谱的发射波长基本一致,大约在856和862 nm左右.在固态下酞菁分子排列紧密,分子间相互作用增强导致了荧光发射波长的巨大红移.
(1) Braun,A.;Tcherniac,J.Dtsch.Ber.Chem.Ges.1907,40,2709.
(2) Ng,D.N.;Jiang,J.Chem.Soc.Rev.1997,26,433.
(3)Lecours,S.M.;Guan,H.W.;Dimagno,S.G.;Wang,C.H.; Therien,M.J.J.Am.Chem.Soc.1996,118,1497.
(4) Priyadarshy,S.;Therien,M.J.;Beratan,D.N.J.Am.Chem. Soc.1996,118,1504.
(5)Toupance,T.;Ahsen,V.;Simon,J.J.Am.Chem.Soc.1994,116, 5352.
(6) Toupance,T.;Benoit,H.;Sarzain,D.;Simon,J.J.Am.Chem. Soc.1997,119,9191.
(7) Moskalev,P.N.;Kirin,I.S.Russ.J.Phys.Chem.1972,46, 1019.
(8) Moskalev,P.N.;Kirin,I.S.Russ.Inorg.Chem.1971,16,57.
(9) Belarbi,Z.;Maitrot,M.;Ohta,K.Chem.Phys.Lett.1988,143, 400.
(10)Toupance,T.;Mineau,P.L.;Simon,J.J.Phys.Chem.1996, 100,11704.
(11) Rosenthal,I.Photochem.Photobiol.1991,53,859.
(12)Wöhrle,D.;Iskandar,N.;Graschew,G.Photochem.Photobiol. 1990,51,351.
(13)Wöhrle,D.;Hirth,A.;Bogdahn-Rai,A;Schnurpfeil,l.Russ. Chem.Bull.1998,47,807.
(14)Cheng,C.H.;Fan,Z.Q.;Yu,S.K.;Jiang,W.H.;Wang,X.;Du, X.G.;Chang,Y.C.;Ma,C.Y.Appl.Phys.Lett.2006,88, 213505.
(15) Sastre,A.;Gouloumis,A.;Vázquez,P.;Torres,T.;Doan,V.; Schwartz,B.J.;Wudl,F.;Echegoyen,L.;Rivear,J.Org.Lett. 1999,1,1807.
(16) Vacus,J.;Simon,J.Adv.Mater.1995,7,797.
(17)Somerauer,M.C.;Rager,M.;Hanack,M.J.Am.Chem.Soc. 1996,118,10085.
(18) Rager,C.;Schmid,G.;Hanack,M.Chem.Eur.J.1999,5,280.
(19)Hanack,M.;Schmid,G.;Sommerauer,M.Angew.Chem.1993, 105,1540.
(20)Hanack,M.;Schmid,D.;Sommerauer,M.Angew.Chem.Int. Edit.1993,32,1422.
(21) Snow,A.W.;Jarvis,N.L.J.Am.Chem.Soc.1984,106,4706.
(22) Baldassare,D.B.Spectroscopy of the Excited State;Plenum Press:New York,1972,p 340.
(23) Huang,Z.Y.;Huang,J.D.;Chen,N.S.;Huang,J.L.Dyes Pigment.2008,77,584.
November 2,2010;Revised:January 4,2011;Published on Web:March 11,2011.
Photoluminescence and Electroluminescence of the Novel Soluble Zinc Phthalocyanine
BAI Qing-Long1,2ZHANG Chun-Hua2CHENG Chuan-Hui1,*LI Wan-Cheng SHEN Ren-Sheng1DU Guo-Tong1,3,*
(1School of Physics and Optoelectronic Technology,Dalian University of Technology,Dalian 116024,Liaoning Province,
P.R.China;2College of Chemistry and Chemical Engineering,Inner Mongglia University for Nationalities,Tongliao 028043, Inner Mongglia,P.R.China;3State Key Laboratory of Integrated Optoelectronics,College of Electronic Science and Engineering, Jilin University,Changchun 130021,P.R.China)
α(β)-tetra-(methoxy-phenoxy)-zincphthalocyanine are synthesized byemploying 3(4)-nitrobenzene-1,2-dicarbonitrile as precursors.They are characterized by spectrum methods and elemental analysis.The UV-Vis spectrum,photoluminescence spectra of spin-coated film and solid pellet are compared.The electroluminescent devices are fabricated by using spin coating.The results indicate that the fluorescence of solid phthalocyanine has a red-shift of more than 145 nm compared to that in solution. The fluorescences are broader in solid state than that in solution.The fluorescence of β-substituted phthalcyanines has a more red-shift than α-substituted phthalcyanines.The electroluminescent spectra around 856 and 862 nm are consisted with the photoluminescence spectra of spin-coated film.The shorter inter-molecule space leads to the large red-shift of the fluorescence.
Soluble phthalocyanine;Synthesis;Photoluminescence;Electroluminescence; Red-shift
O644
∗Corresponding authors.CHENG,Chuan-Hui,Email:chengch79@yahoo.cn;Tel:+86-13591833580,+86-13504306097.
DU Guo-Tong,Email:dugt@dlut.edu.cn;Tel:+86-13840838794,+86-13019136698.
The project was supported by the National Natural Science Foundation of China(60807009),Fundamental Research Funds for the Central Universities,China and Specialized Research Fund for the Doctoral Program of Higher Education,China(200801411038).
国家自然科学基金(60807009),中央高校基本科研业务费专项资金和高等学校博士学科点专项科研基金(200801411038)资助项目
猜你喜欢
纺织科学研究(2023年12期)2023-12-19 12:36:08
纺织科学研究(2020年4期)2020-04-30 05:37:42
安徽化工(2018年4期)2018-09-03 07:11:48
中央民族大学学报(自然科学版)(2016年2期)2016-06-27 01:29:06
湖北师范大学学报(自然科学版)(2015年1期)2016-01-10 08:41:29
浙江理工大学学报(自然科学版)(2015年7期)2015-03-01 02:54:21
Journal of Southeast University(English Edition)(2014年4期)2014-09-06 10:49:51
应用化工(2014年10期)2014-08-16 13:11:29
无机化学学报(2014年4期)2014-02-28 17:31:11
无机化学学报(2014年1期)2014-02-28 17:30:11
