
四齿配体[Ru(iph)(L)2]2+(L=cpy,mpy,npy)配合物的结构和光谱特征
2011-11-30 10:42:04张建坡张红星
物理化学学报 2011年5期
张建坡 金 丽,* 张红星
(1吉林化工学院,化学与制药工程学院,吉林吉林132022; 2吉林大学理论化学研究所,理论化学计算国家重点实验室,长春130023)
四齿配体[Ru(iph)(L)2]2+(L=cpy,mpy,npy)配合物的结构和光谱特征
张建坡1金 丽1,*张红星2
(1吉林化工学院,化学与制药工程学院,吉林吉林132022;2吉林大学理论化学研究所,理论化学计算国家重点实验室,长春130023)
采用密度泛函的B3LYP和UB3LYP方法分别优化了一系列[Ru(iph)(L)2]2+(L=cpy(1),mpy(2),npy
(3);其中iph为2,9-双(1′-甲基-2′-咪唑)-1,10-邻二氮杂菲,cpy为4-氰基嘧啶,mpy为4-甲基嘧啶,npy为4-氮二甲基嘧啶)配合物的基态和激发态结构.利用含时密度泛函理论(TD-DFT)方法,结合极化连续介质(PCM)模型计算了它们在丙酮溶液中的吸收和发射光谱.研究结果表明:优化得到的几何结构参数和相应的实验值符合得非常好.1和2的最高占据分子轨道主要由金属的d轨道和iph配体的π轨道构成,但是3主要占据在npy配体上,而它们的最低空轨道主要由iph配体的π反键轨道占据.因此,1和2的最低能吸收和发射属于金属到配体(MLCT)和配体内部(ILCT)的电荷转移跃迁,而3属于两个配体之间的电荷转移(LLCT)跃迁.三个配合物的最低能吸收分别在509 nm(1),527 nm(2)和563 nm(3),其磷光发射分别在683 nm(1),852 nm(2)和757 nm
(3).这显示出通过调节L配体的π电子给予能力可以改变最低能吸收和发射的跃迁性质和发光颜色.
四齿配体Ru配合物;激发态;含时密度泛函理论;光谱特征;电荷转移
1 引言
钌配合物具有丰富的光物理和光化学性质,并在光电转换和生物等领域内有着广泛的应用.1-3最早的标志性钌配合物是[Ru(bpy)3]2+(bpy=2,2′-bipyridine)及其衍生物.4-6在水溶液中,[Ru(bpy)3]2+配合物在452和426 nm处产生强度很大的金属到配体的跃迁(MLCT);在352和320 nm处产生了发生在金属中心(MC)的d→d跃迁;另外在287 nm出现了很强的π→π*吸收谱.由于[Ru(bpy)3]2+具有光诱导电荷分离功能,使其广泛地应用于光敏剂、光催化、太阳能电池和光致变色等领域,从而拉开了对这类配合物研究的序幕.
1993年,Grätzel小组7-12合成了一系列的[Ru(bpy)3]2+衍生物RuL2X2,其中L为双齿配体,X为单齿配体.这类配合物除具有优良的光谱性能外,还存在顺式和反式两种结构,反式结构存在较小的空间位阻,相对较稳定,但在一定情况下二者可以转换.于是Grätzel小组率先采用原子将中间的两个双齿配体连接起来,构成一个大的四齿桥联配体,但由于桥联配体不够稳定,故得到的这种物质不是很理想,需要进一步改进.12,13
最近,Thummel小组14,15从稳定的四齿配体出发,得到了这种RuLX2配合物,L为一个稳定的四齿配体,如2,9-双嘧啶-1,10-邻二氮杂菲,2,9-双(1′-甲基-2′-咪唑)-1,10-邻二氮杂菲等,X为4-取代嘧啶.通过对单齿和四齿配体改造发现,改造四齿配体对结构和光谱性质的影响不如改造单齿配体更加明显.为了深入地剖析这类配合物的结构和光谱性质,以及单齿配体上不同取代基对其结构和光谱的重要影响,本文采用密度泛函方法,16-19完成了对[Ru(iph) (L)2]2+(L=cpy(1),mpy(2),npy(3);其中iph为2,9-双(1′-甲基-2′-咪唑)-1,10-邻二氮杂菲,cpy为4-氰基嘧啶,mpy为4-甲基嘧啶,npy为4-氮二甲基嘧啶)配合物的结构和光谱特征的系统理论研究.研究表明,单齿配体的4-取代位置对配合物的影响巨大,强的给电子取代基甚至可以使最低能吸收和发射的跃迁性质发生根本转变.
2 计算方法
配合物(1-3)在基态和激发态均采用C2对称性.为了得到更为合理的几何结构,以配合物(2)为例选择不同的基组和方法分别优化,所涉及的优化几何参数列在表1中.方法选择采用从头算的MP2方法20和密度泛函的MPW1PW91,BP86和B3LYP方法,16-19优化结果显示出密度泛函的B3LYP方法得到的键长和键角更接近于实验值.基组选择中,对金属分别采用LANL2DZ和SDD基组,对其他原子分别采用6-31G和6-31G*基组,发现金属采用SDD基组更为合理,而其他原子采用两种基组优化得到的结果基本一致.考虑节省计算成本,本实验采用B3LYP/SDD-6-31G方法,并采用选定的方法优化了配合物1和3的基态结构.
在优化基态结构的基础上,采用UB3LYP方法分别优化了1-3最低能三重激发态结构.根据Franck-Condon垂直跃迁原理,利用含时密度泛函(TDDFT)21结合PCM溶剂化模型22计算了1-3在丙酮溶液中的吸收和发射光谱.以上计算通过执行Gaussian 03程序包23在origin/3900服务器上完成.
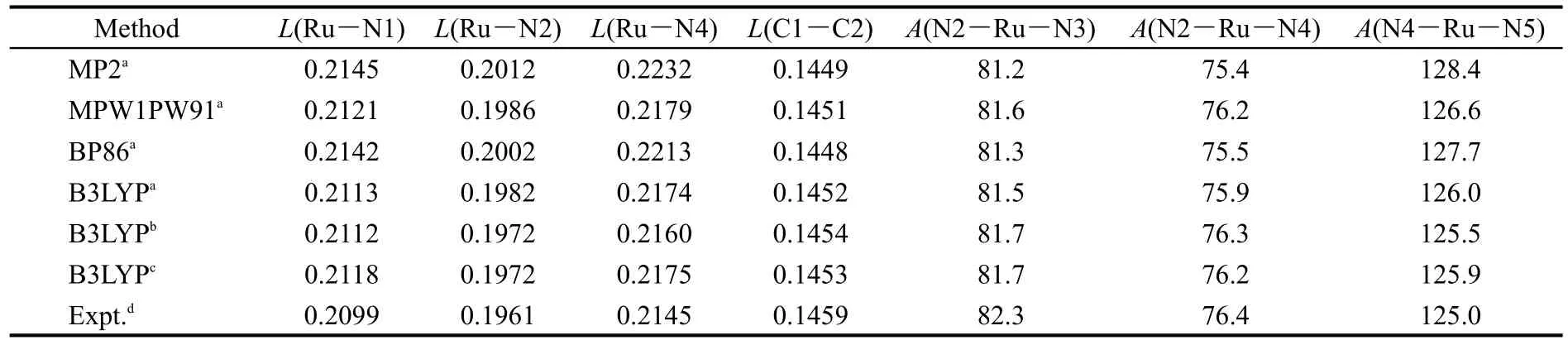
表1 采用不同的方法和基组优化得到的配合物[Ru(iph)(L)2]2+(2)的几何参数Table 1 Optimized geometry parameters of complex[Ru(iph)(L)2]2+(2)with the different methods and basis sets
3 结果和讨论
3.1 基态和激发态结构
图1给出了1-3的结构简图,其基态几何参数以及2相应的实验值列于表2中.由表2知,三个配合物都具有1A基态和3A激发态.2基态的三个重要的Ru-N键长与实验值分别相差了0.0013、0.0011和0.0015 nm,而C1-C2键长则几乎与实验值相等,且三个键角的理论值与实验值之差也都在0.6°范围内,以上数据均说明计算结果准确、可靠.值得注意的是,二面角N2-C2-C1-N4为-2.4°,表明四齿配体并不完全在同一个平面内,由于空间效应的影响,四齿配体平面发生了较小的扭曲.其扭曲的角度要小于trans-Ru(bpy)2X2配合物,8,9表明近赤道平面有一个稳定的四齿配体,比两个双齿配体所受的空间位阻更小,结构更加稳定.1和3也有相似的几何结构,表明更换R取代基对其主体几何结构影响不大.
表2中还列出了配合物激发态的几何参数.与基态相比,三个配合物的Ru-N1和Ru-N2键长分别增加了约0.0025和0.0100 nm,增加较明显,而Ru-N4键长则略有缩短(0.0006-0.0020 nm).激发态的N2-Ru-N3和N2-Ru-N4键角变小,而N4-Ru-N5键角变大.表明电子跃迁必然发生在两种配体上,从而消弱了Ru原子与1,10-邻二氮杂菲部分的相互作用,增强了其与咪唑部分的相互作用,其对应着1-3最低能吸收和发射必然包含有部分π-π*跃迁.此外,激发态N2-C2-C1-N4二面角变大,显示出四齿平面在激发态时受空间效应影响进一步扭曲.

图1 配合物1-3的结构简图及原子编号Fig.1 Structure scheme and atom-numbering of complexes 1-3
3.2 配合物在丙酮溶液中的吸收光谱
在基态几何基础上,利用TD-DFT方法计算了1-3在丙酮溶液中的紫外-可见吸收光谱,并在图2中给出了模拟的Gaussian型吸收曲线.将1-3中较强、典型吸收列于表3中,并把吸收中组态系数最大跃迁的始态和终态分子轨道成份及能量表示在图3中.
从图2、表3和图3可以看出,1-3都有三个明显的吸收带,它们的最低吸收能分别在509 nm(1)、527 nm(2)和563 nm(3).1在509 nm的吸收来自于分子轨道74b→77a的激发,分子轨道74b为最高占据分子轨道(HOMO),由d(Ru)和π(iph)轨道成份构成;而77a是最低空轨道(LUMO),主要由配体iph的π反键轨道及少量的金属d轨道占据.因此,此跃迁是属于[d(Ru)+π(iph)→d(Ru)+π*(iph)]的MLCT,以及发生在iph配体内部的电荷转移(ILCT)跃迁.与1类似,2的最低能吸收也是来自最高占据分子轨道(HOMO)到最低未占据分子轨道(LUMO)的跃迁,并且具有相似的跃迁性质.值得注意的是,配合物3的最低能吸收来自于分子轨道80b→83a,分子轨道80b主要占据在配体的π(npy)轨道上,而83a属于iph配体的π*轨道,导致其跃迁性质变为配体到配体的电荷转移(LLCT)跃迁.比较1-3最低能吸收可以看出,随着嘧啶配体上R取代基的给电子能力逐渐增强(1<2<3),最低能吸收波长发生明显的红移,并且使跃迁性质发生明显的改变(MLCT/ILCT,变为LLCT).由图3可知,随着R取代基的给电子能力逐渐增强,HOMO和LUMO轨道能都有所升高,只是HOMO轨道能升高的更多,导致其能级间隙变小,波长红移.通常来讲,最低能吸收有较高比例的MLCT跃迁会使发射的磷光性能比较突出,而配合物3的最低能吸收变为LLCT跃迁,会导致其最低能发射可能具有磷光和荧光双重性能.实验上也观测到了2的最低能吸收在536 nm,这与计算得到的527 nm符合得非常好.
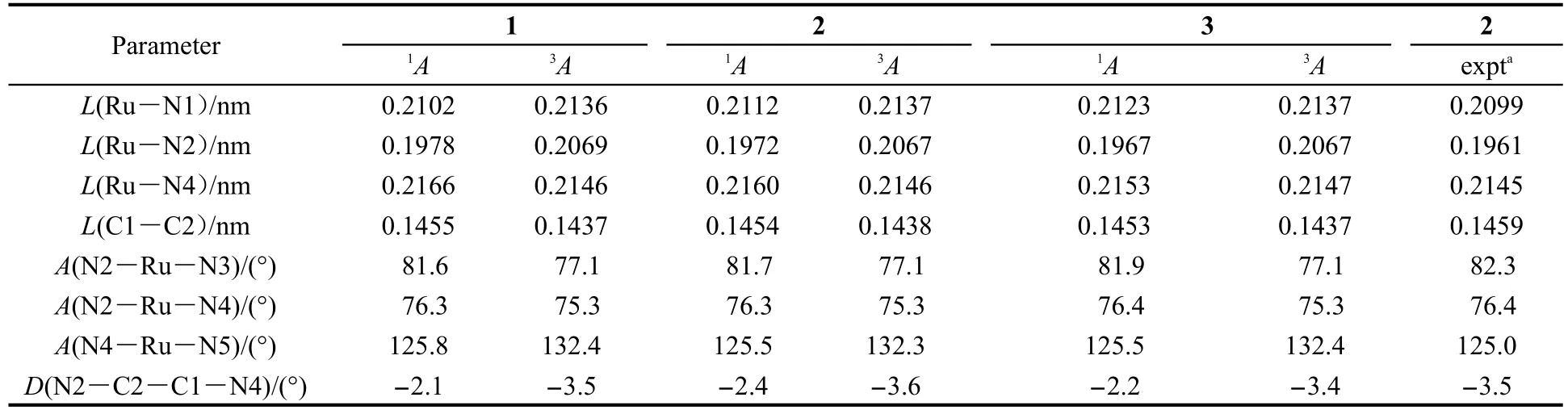
表2 1-3的基态和三重激发态几何结构参数及其实验值Table 2 Optimized geometry parameters of 1-3 in the ground and triplet excited states,and the experimental values

图2 配合物1-3在丙酮溶液中模拟的Gaussian型吸收曲线Fig.2 Simulated absorption spectra with Gaussian curves based on the data calculated in acetone for complexes 1-3f:oscillator strength
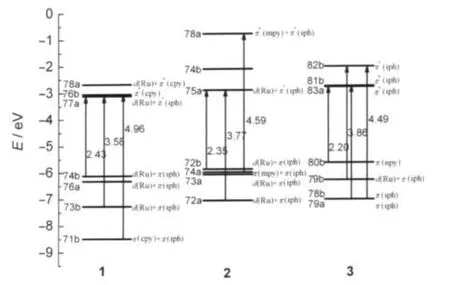
图3 TD-DFT计算得到配合物1-3的相关轨道能级图Fig.3 Orbital energy level diagrams involved for 1-3 under TD-DFT calculations
1-3的强吸收分别在345 nm(1)、329 nm(2)和321 nm(3),其振子强度分别为0.3804、0.3546和0.2184,如此强的吸收应该是实验中最容易观察到的.类似于上面的分析,这三个吸收也都与金属和iph配体有关(表3和图3),它们都属于MLCT/ILCT跃迁.从图2可以直观看出,随着给电子能力的增强,强吸收峰有比较小的蓝移.
三个配合物高能吸收带代表性吸收分别在250 nm(1)、270 nm(2)和276 nm(3),它们也具有较大的振子强度(3达到了0.5760).配合物1和2的此吸收主要发生在单齿配体上,属于单齿配体内部的电荷转移跃迁(ILCT),并伴有少量的两个配体之间跃迁的微扰(LLCT).但是3的此跃迁主要发生在四齿配体内部,虽都属于ILCT跃迁,但有本质的区别.从图2也可以直观的看出,随着给电子能力的增强,高能吸收峰有小的红移,这与最低能吸收的红移趋势一致.
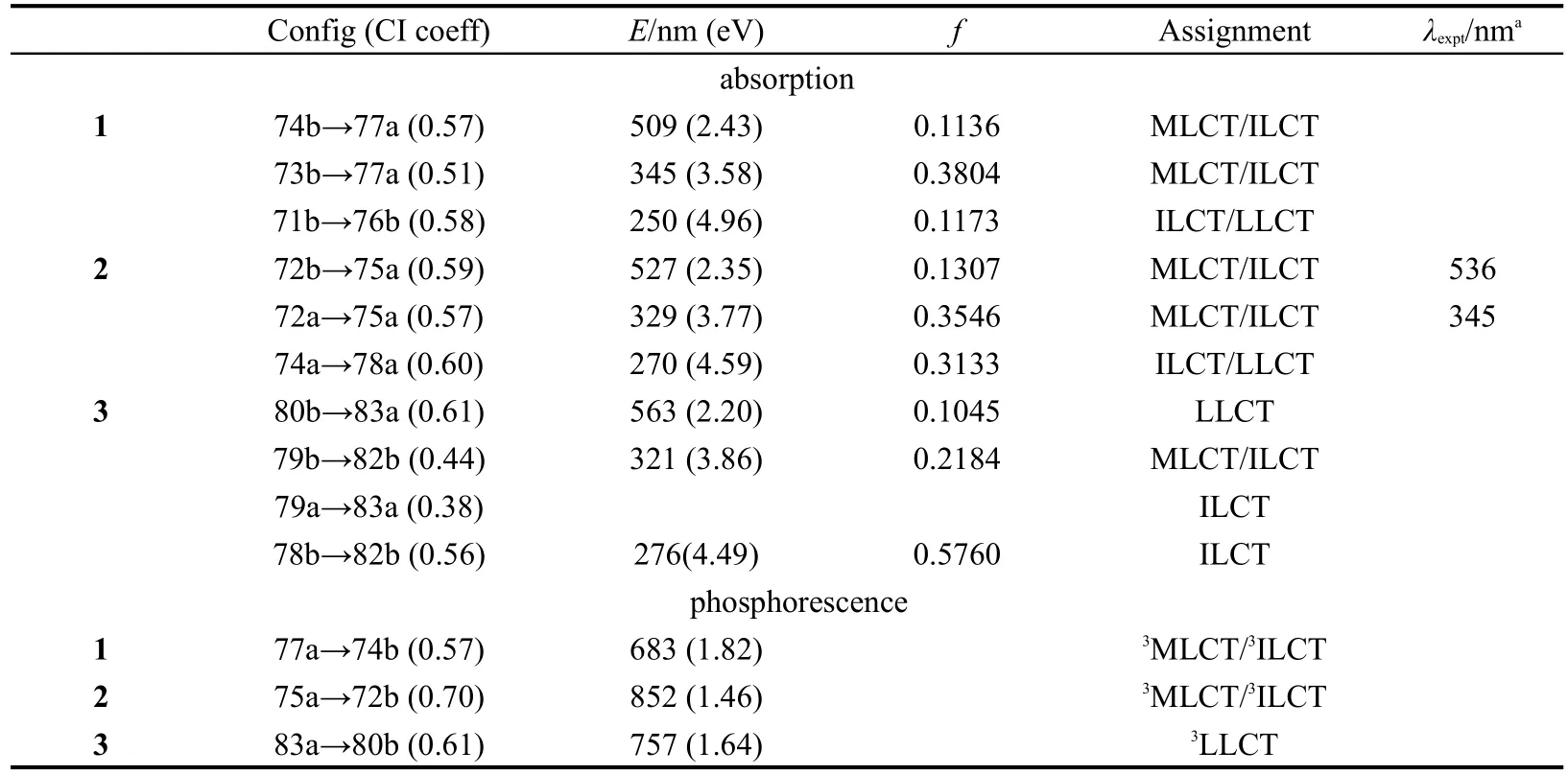
表3 1-3在丙酮溶液中的吸收和磷光发射数据及其实验值Table 3 Absorption and phosphorescence of 1-3 in acetone under TD-DFT calculations and the experimental values
3.3 配合物在丙酮溶液中的磷光发射光谱
表3还列出了1-3在丙酮溶液中最低能发射的磷光光谱数据.为了直观地理解配合物的跃迁特征,在图4中给出了在最低能发射中具有最大组态系数的电子跃迁图.
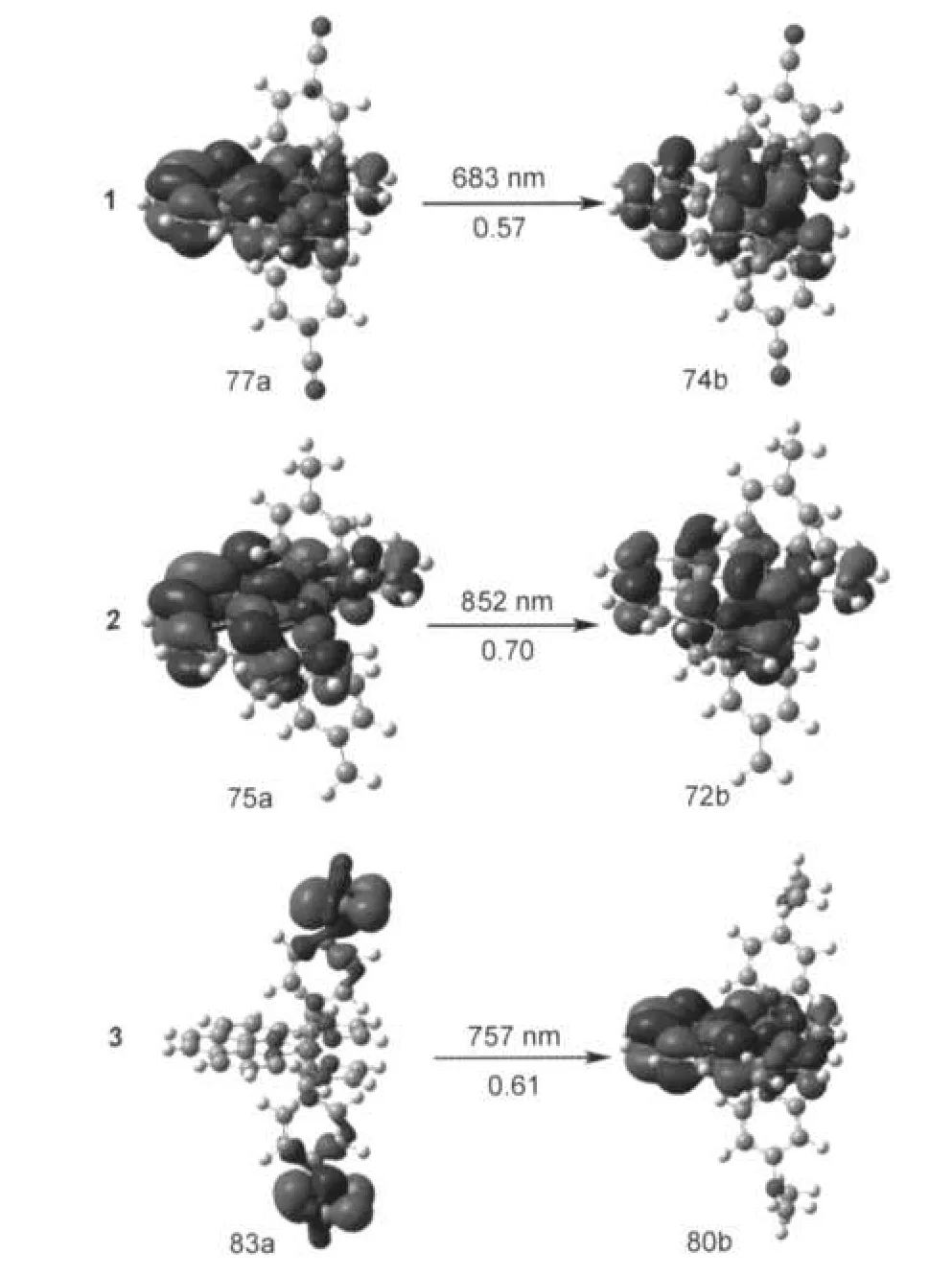
图4 配合物1-3在丙酮溶液中最低能发射的最大CI系数的跃迁分子轨道图形Fig.4 The molecular orbital graphics with the maximum CI coefficients for the lowest energy emissions of complexes 1-3 in acetone
由表3和图4可知,三个配合物的磷光发射分别在683 nm(1),852 nm(2)和757 nm(3).1在683 nm处的发射来自于分子轨道77a→74b的激发,分子轨道74b由Ru的d轨道和iph配体的π轨道构成,而77a主要占据在iph配体的π反键轨道及少量的金属d轨道上,因此这个跃迁具有3MLCT/3ILCT特征,这与其最低能吸收在跃迁性质方面是一致的.与1类似,2的发射也与其最低能吸收有相似的3MLCT/3ILCT特征.与1和2相比,3的最低能发射在757 nm,属于3LLCT特征,这和许多有机金属配合物以3MLCT跃迁为主有很大的不同,24,253虽具有较大的给电子基团,但是最低能发射波长并没有发生红移.这主要是由于,跃迁性质的转变导致其最低能发射具有荧光和磷光双重性能,这个规律在一类Os(II)(CO)3(tfa)(acac(X)2)配合物26中也有发现,并且计算得到其最低能荧光发射在621 nm处.
比较1-3的最低能吸收和发射,配合物1和2有相似的轨道成份和跃迁性质,但与3有本质的差别,它们的能量差分别为0.61 eV(1),0.89 eV(2)和0.56 eV(3),这些适当的斯托克斯频移和它们基态到激发态结构小的改变是一致的.3的斯托克斯频移(0.56 eV)最小,这和轨道分析中得到的最低能发射具有磷光和荧光双重特征相对应.另外,通过它们的最低能发射的分子轨道图(图4)可以进一步理解三个配合物的分子轨道占据及跃迁情况.通过以上研究发现,此类配合物对嘧啶配体的4-取代位置很敏感,强的给电子取代基可以使最低能吸收和发射的跃迁性质发生改变,使发射具有双重特征.
4 结论
从理论上研究了三个不同R取代基的四配位配体[Ru(iph)(L)2]2+配合物的几何结构和光谱特征.计算结果显示出,优化得到的几何结构参数及其计算得到的光谱数据与相应的实验值符合得非常好.当R为吸电子(CN)或者较弱给电子(CH3)取代基时,这类配合物的最低能吸收和发射都来自于MLCT/ ILCT跃迁.而当R为强的给电子取代基(N(CH3)2)时,最低能吸收和发射的跃迁特征变为LLCT,从而使其发射具有磷光和荧光双重特征.这表明,此类配合物对单齿配体嘧啶的4-取代位置非常敏感,通过调节4-取代位置上的R取代基可以改变它们最低能吸收和发射的跃迁性质.此外,后续的工作调整单齿和四齿配体对结构和光谱的影响,也将继续展开.希望通过此理论研究给实验合成以指导,以便开发出更多优良的发光材料.
(1) Maubert,B.;McClenaghan,N.D.;Indelli,M.T.;Campagna,S. J.Phys.Chem.2003,107,447.
(2) Bergmini,G.;Saudan,C.;Ceroni,P.;Maestri,M.;Balzani,V.; Gorka,M.;Lee,S.K.;Heyst,J.;Vögtle,F.J.Am.Chem.Soc. 2004,126,16466.
(3)Wong,C.Y.;Tong,G.S.M.;Che,C.M.;Zhu,N.Angew.Chem. Int.Edit.2006,45,2694.
(4) Juris,A.;Balzani,V.;Barigelletti,F.;Campagna,S.;Belser,P.; Zelewski,A.Coord.Chem.Rev.1988,84,85.
(5) Klassen,D.M.;Crosby,G.A.J.Chem.Phys.1965,43,1498.
(6) Lytle,F.E.;Hercules,D.M.J.Am.Chem.Soc.1969,91,23.
(7)Nazeeruddin,M.K.;Kay,A.;Rodicio,I.;Humphry-Baker,R.; Müeller,E.;Liska,P.;Vlachopoulos,N.;Grätzel,M.J.Am. Chem.Soc.1993,115,6382.
(8) Nazeeruddin,M.K.;Péchy,P.;Renouard,T.;Zakeeruddin,S. M.;Humphry-Baker.;Comte,R.P.;Liska,P.;Cevey,L.;Costa, E.;Shklover,V.;Spiccia,L.;Deacon,G.B.;Bignozzi,C.A.; Grätzel,M.J.Am.Chem.Soc.2001,123,1613.
(9) Zakeeruddin,S.M.;Nazeeruddin,M.K.;Humphry-Baker,R.; Péchy,P.;Quagliotto,P.;Barolo,C.;Visvardi,G.;Grätzel,M. Langmuir 2002,18,952.
(10) Nazeeruddin,M.K.;Angelis,F.D.;Fantacci,S.;Selloni,A.; Viscardi,G.;Liska,P.;Ito,S.;Taleru,B.;Grätzel,M.J.Am. Chem.Soc.2005,127,16835.
(11)Wang,Z.S.;Yamaguchi,T.;Sugihara,H.;Arakawa,H. Langmuir 2005,21,4272.
(12) Barolo,C.;Nazeeruddin,M.K.;Fantacci,S.;Censo,D.D.; Comte,P.;Liska,P.;Viscard,G.;Quagliotto,P.;Angelis,F.D.; Ito,S.;Grätzel,M.Inorg.Chem.2006,45,4642.
(13) Renouard,T.;Fallahpour,R.A.;Nazeeruddin,M.K.;Humphry-Baker,R.;Gorelsky,S.I.;Lever,A.B.P.;Grätzel,M.Inorg. Chem.2002,41,367.
(14)Zong,R.;Thummel,R.P.J.Am.Chem.Soc.2004,126,10800.
(15) Zhang,G.;Zong,R.F.;Tseng,H.W.;Thummel,R.P.Inorg. Chem.2008,47,990.
(16) Hohenberg,P.;Kohn,W.Phys.Rev.1964,136,B864.
(17) Kohn,W.;Sham,L.J.Phys.Rev.1965,140,A1133.
(18) Becke,A.D.J.Chem.Phys.1993,98,5648.
(19) Lee,C.;Yang,W.;Parr,R.G.Phys.Rev.B 1988,37,785.
(20) Møller,C.;Plesset,M.S.Phys.Rev.1934,46,618.
(21) Casida,M.E.;Jamorski,C.;Casida,K.C.;Salahub,D.R. J.Chem.Phys.1998,108,4439.
(22) Cossi,M.;Scalmani,G.;Regar,N.;Barone,V.J.Chem.Phys. 2002,117,43.
(23) Frisch,M.J.;Trucks,G.W.;Schlegel,H.B.;et al.Gaussian 03, Revision C.02;Gaussian,Inc.:Wallingford,CT,2004.
(24)Zhou,X.;Zhang,H.X.;Pan,Q.J.;Li,M.X.;Wang,Y.;Che,C. M.Eur.J.Inorg.Chem.2007,15,2181.
(25) Li,M.X.;Zhou,X.;Pan,Q.J.;Zhang,H.X.;Fu,H.G.;Sun,C. C.Chem.J.Chin.Univ.2007,28,2377.[李明霞,周 欣,潘清江,张红星,付宏刚,孙家锺.高等学校化学学报,2007, 28,2377.]
(26) Zhang,J.P.;Zhou,X.;Bai,F.Q.;Zhang,H.X.;Tang,A.Q. Theor.Chem.Acc.2009,122,31.
January 3,2011;Revised:February 10,2011;Published on Web:March 25,2011.
Structures and Spectroscopic Properties of[Ru(iph)(L)2]2+(L=cpy,mpy, npy)Complexes Containing Tetradentate Ligands
ZHANG Jian-Po1JIN Li1,*ZHANG Hong-Xing2
(1School of Chemical and Pharmaceutical Engineering,Jilin Institute of Chemical Technology,Jilin 132022,Jinlin Province,P.R. China;2State Key Laboratory of Theoretical and Computational Chemistry,Institute of Theoretical Chemistry, Changchun 130023,P.R.China)
The geometries of ground and excited states of a series of ruthenium complexes[Ru(iph)(L)2]2+(L=cpy(1),mpy(2),npy(3);iph=2,9-di(1-methyl-2-imidazole)-1,10-phenanthroline,cpy=4-cyano pyridine, mpy=4-methyl pyridine,npy=4-N-methyl pyridine)were optimized by the Becke′s three-parameter functional and the Lee-Yang-Parr(B3LYP)functional and unrestricted B3LYP methods,respectively.Timedependent density functional theory(TD-DFT)method at the B3LYP level together with the polarized continuum model(PCM)were used to obtain their absorption and phosphorescent emission spectra in acetone media based on their optimized ground and excited-state geometries.The results revealed that the optimized structural parameters agreed well with the corresponding experimental results.The highest occupied molecular orbitals were localized mainly on the d orbital of the metal and the π orbital of the iph ligand for 1 and 2,and the npy ligand for 3,while the lowest unoccupied molecular orbitals were mainly composed of π*orbital of the iph ligand.Therefore,the lowest-lying absorptions and emissions were assigned to the metal to ligand charge transfer(MLCT)/intra-ligand charge transfer(ILCT)transition for 1 and 2,and the ligand to ligand charge transfer(LLCT)transition for 3.The lowest-lying absorptions are at 509 nm (1),527 nm(2),and 563 nm(3)and the phosphorescence emissions at 683 nm(1),852 nm(2),and 757 nm (3).The calculation results show that the absorption and emission transition characteristics and the phosphorescence color can be changed by altering the πelectron-donating ability ofthe Lligand.
Tetradentate ligands Ru complexes;Excited state;Time-dependent density functional theory;Spectroscopic property;Charge transfer
O641
*Corresponding author.Email:canoe8013@126.com;Tel:+86-432-63083022.
The project was supported by the Foundation of State Key Laboratory of Theoretical and Computational Chemistry,China.
理论化学计算国家重点实验室开放课题基金资助项目
猜你喜欢
数字制造科学(2021年3期)2021-09-27 01:40:06
机械研究与应用(2021年4期)2021-09-15 05:41:56
汕头大学学报(自然科学版)(2020年4期)2020-12-14 07:04:58
汕头大学学报(自然科学版)(2020年4期)2020-12-14 07:04:58
制造业自动化(2019年7期)2019-07-26 09:25:38
World Journal of Diabetes(2019年7期)2019-07-23 11:52:08
分析化学(2017年12期)2017-12-25 12:03:53
分析化学(2017年12期)2017-12-25 06:58:03
露天采矿技术(2017年5期)2017-06-05 15:06:16
原子与分子物理学报(2015年3期)2015-11-24 12:49:36
