
L-苯甘氨酸衍生物的合成及其抗糖尿病活性*
2011-11-27 04:44李华冲晏菊芳苏小燕孔令强杨大成
合成化学 2011年4期
李华冲,晏菊芳, 苏小燕, 孔令强, 杨大成
(1. 西南大学 化学化工学院,重庆 400715; 2. 成都地奥制药集团 药物筛选中心,四川 成都 610041)
选用价廉易得的天然原料,采用相对简单的合成方法制备结构新颖的抗糖尿病药物,是化学与药学工作者的艰巨任务。
苯甘氨酸衍生物用途广泛,如D-对羟基苯甘氨酸是β-内酰胺类半合成抗生素的侧链,用于生产如阿莫西林、头孢羟氨苄等抗生素药物[1];氯代苯甘氨酸衍生物不仅用于抗菌类药物、抗血栓类药物以及农药等的制备,而且还用于青霉素类衍生物、头孢菌素类衍生物的合成[2]等。但到目前为止,苯甘氨酸及其衍生物作为抗糖尿病药物的研究还未见文献报道。
本文借鉴具有强的PPARγ激动活性的L-酪氨酸衍生物的结构特征[3],设计了L-间氨基苯甘氨酸作为关键中间体,通过对其结构的进一步修饰制得具有多种结构特点的L-间氨基苯甘氨酸衍生物,经过抗糖尿病活性筛选,期望得到具有抗糖尿病活性的先导化合物。
本文以L-苯甘氨酸为原料,通过苯环硝化[4~6]、α-氨基保护[4,5,7~9]、羧基保护[10]及硝基还原[11~14]等反应,合成了中间体L-间硝基苯甘氨酸衍生物(1,2a~2c和5c),产物L-间氨基苯甘氨酸(4)和L-间氨基苯甘氨酸衍生物(3a~3c,6c)(Scheme 1),其中2b,5c, 3b,3c及6c为新化合物,其结构经1H NMR,13C NMR及MS表征。初步测试了部分化合物的抗糖尿病活性。




CompabcRBocFmocCbz
Scheme1
1 实验部分
1.1 仪器与试剂
X-6型精密显微熔点仪;WZZ-2S型自动旋光仪;AV-300型超导核磁共振仪(DMSO-d6为溶剂,TMS为内标);GX型FT-IR红外光谱仪(KBr压片);Agilent 1946B MS(ESI MS)型质谱仪。
1[6]按文献方法合成;L-苯甘氨酸(L-Phg),苄基琥珀酰亚胺基碳酸酯(Z-OSu),二叔丁氧基碳酸酐(Boc2O)和9-芴甲基丁二酰亚胺基碳酸酯(Fmoc-OSu),分析纯,成都凯泰新技术有限责任公司;其余所用试剂均为市售分析纯。
1.2 合成
(1) 2的合成通法[8,9]
在反应瓶中加入1 2 mmol, 10%Na2CO3溶液或10%K2CO3溶液5 mL,冰浴冷却下搅拌使其溶解;缓慢滴加Fmoc-OSu 1.96 mmol(或Z-OSu 2.2 mmol,或Boc2O 2.4 mmol)的丙酮(3.5 mL)溶液,滴毕,于20 ℃~30 ℃反应至终点(TLC跟踪,pH 9~10)。加适量水稀释,分液,水层用乙醚(3×5 mL)萃取。冰浴冷却下,水相用2 mol·L-1HCl调至pH 3~4,减压蒸除丙酮,残余物用乙酸乙酯(3×10 mL)萃取,合并萃取液,用饱和NaCl溶液洗至中性,无水MgSO4干燥,减压蒸除乙酸乙酯得2a~2c。
2a: 淡黄色固体,收率83.2%,m.p.143 ℃~150 ℃,[α]+72°(c1.0, CH3OH)。
2b: 类白色固体,收率87.4%,m.p.101.4 ℃~105.6 ℃,[α]+43.8°(c2.4, DMF);1H NMRδ: 4.24~4.32(m, 1H, CH), 4.35(s, 2H, CH2), 5.45(d,J=7.95 Hz, 1H, CH), 7.28~7.35(m, 2H, ArH), 7.39~7.44(m, 2H, ArH), 7.68(d,J=7.65 Hz, 1H, ArH), 7.75(d,J=7.20 Hz, 2H, ArH), 7.88~7.90(m, 3H, ArH, CONH), 8.20(d,J=7.83 Hz, 1H, ArH), 8.36(s, 1H, ArH), 8.47(d,J=8.04 Hz, 1H, ArH), 13.24(s, 1H, CO2H);13C NMRδ: 46.6, 57.2, 66.0, 120.1, 122.4, 122.9, 125.3, 127.1, 127.7, 130.0, 134.8, 139.8, 140.8, 143.7, 143.8, 147.8, 155.9, 171.2; ESI-MSm/z: 441.2{[M+Na]+}。
2c: 黄色固体,收率97.9%,m.p.72.5 ℃~74.3 ℃,[α]+122.0°(c2.0, CHCl3); ESI-MSm/z: 331.2{[M+H]+}。
(2) 5c的合成[10]
在反应瓶中加入2c5 mmol和乙酸乙酯10 mL,搅拌使其溶解;滴加三乙胺0.7 mL和苄溴5 mmol,滴毕,于75 ℃回流反应至终点(TLC跟踪)。过滤,滤饼用乙酸乙酯(3×5 mL)洗涤,合并滤液与洗液,依次用5%NaHCO3溶液、饱和NaCl溶液各洗两次,用无水MgSO4干燥,减压蒸除溶剂,残余物用混合溶剂[V(乙酸乙酯)∶V(石油醚)=1∶3]重结晶得白色固体5c,收率86.2%,m.p.120.1 ℃~123.6 ℃;1H NMRδ: 5.03(s, 2H, CH2), 5.06(s, 2H, CH2), 5.51(d,J=6.0 Hz, 1H, CH), 6.08(d,J=3.0 Hz, 1H, CONH), 7.18~7.71(m, 12H, ArH), 8.19~8.22(m, 2H, ArH);13C NMRδ: 57.2, 65.9, 66.7, 122.7, 123.1, 127.7, 127.8, 127.9, 128.1, 128.4, 130.1, 134.9, 135.6, 136.7, 139.6, 147.7, 155.9, 169.8; ESI-MSm/z: 421.0{[M+H]+}, 443.0{[M+Na]+}。
(3) 3,4和6c的合成通法
方法一(Zn/NH4Cl/HOAc还原法): 在圆底烧瓶中加入Zn粉8.9 mmol,NH4Cl 0.2 mmol,2 1.0 mmol及80%乙醇20 mL,搅拌下于65 ℃反应0.5 h~1.0 h;加冰乙酸4.0 mmol,于65 ℃反应至终点(TLC监测)。过滤,滤饼依次用饱和Na2CO3溶液(1×5 mL),饱和NaCl溶液(3×5 mL)洗涤。后处理方法:①滤液用氨水调至pH 5~6,有大量淡黄色沉淀析出,抽滤,滤饼干燥得黄色固体4; ②滤液用饱和Na2CO3溶液调至pH 9~10后用乙醚(3×10 mL)萃取,水层加入乙酸乙酯20mL,在冰浴下用2 mol·L-1HCl调至pH 3~4,用乙酸乙酯萃取(3×10 mL),合并有机层,用饱和NaCl溶液(3×5 mL)洗涤,无水MgSO4干燥,减压蒸除乙酸乙酯得3和6。
方法二(Zn/CaCl2还原法): 参照文献[12]方法合成3,4和6c。
3a: 淡黄色固体,收率82.5%,m.p.109 ℃~120 ℃,[α]+86°(c1.0, CH3OH); ESI-MSm/z: 319.2{[M+Na]+}。
3b: 淡黄色固体,收率85.5%,m.p.124.3 ℃~130.8 ℃,[α]+89.5°(c2.0, DMF);1H NMRδ: 4.23~4.29(m, 3H, CH, CH2), 4.90~4.95(m, 1H, CH), 6.50~7.90(m, 12H, ArH), 8.09(s, 1H, CONH);13C NMRδ: 46.6, 58.4, 66.0, 113.3, 113.6, 115.1, 120.1, 123.6, 125.6, 127.1, 127.7, 128.7, 129.0, 137.5, 140.7, 143.8, 143.9, 143.8, 148.6, 148.8, 155.9, 172.3, 172.9; IRν: 3 446(CONH), 3 307(NH2), 1 687(C=O), 1 604,1 496(C=C) cm-1; ESI-MSm/z: 389.2{[M+H]+},411.1{[M+Na]+}。
3c: 淡黄色固体,收率85.8%,m.p.74.3 ℃~80.6 ℃,[α]+81.36°(c2.2, DMF);1H NMRδ: 4.68(s, 1H, CH), 4.90(s, 2H, NH2), 5.03(s, 2H, CH2), 6.38(d,J=7.50 Hz, 2H, ArH), 6.43~7.34(m, 7H, ArH);13C NMRδ: 57.9, 65.5 113.6, 115.0, 121.5, 127.7, 127.8, 128.4, 129.0, 137.0, 137.5, 151.9, 155.9, 172.2; ESI-MSm/z: 301.1{[M+H]+}, 323.1{[M+Na]+}。
4: 淡黄色固体,收率85.4%,m.p.194.5 ℃~203.4 ℃,[α]+31.8°(c1.1, 2 mol·L-1HCl)。
6c: 白色固体,收率81.8%,m.p.59.8 ℃~64.5 ℃;1H NMRδ: 5.06(s, 2H, CH2), 5.09(s, 2H, CH2), 5.12~5.14(m, 1H, CH), 5.16(s, 2H, NH2), 6.51(d,J=7.60 Hz, 2H, ArH), 6.58(s, 1H, ArH), 6.95~7.00(m, 1H, ArH), 7.26~7.35(m, 10H, ArH), 8.20(d,J=7.50 Hz, 1H, CONH);13C NMRδ: 58.5, 65.7, 66.1, 113.3, 113.8, 115.0, 127.6, 127.8, 127.9, 128.0, 128.4, 129.1, 135.9, 136.5, 137.0, 149.0, 156.0, 170.9; ESI-MSm/z: 391.1{[M+H]+}, 413.0{[M+Na]+}。
1.3 PPAR反应元件(PPRE)激动活性检测
将HepG2肝癌细胞常规培养于37 ℃,5%CO2,含100 U·mL-1链霉素和青霉素的低糖DMEM中,在1.5×104个/孔接种于96孔板后培养过夜,参照转染试剂说明书进行质粒转染。转染的质粒包括带有PPRE和萤火虫荧光素酶报告基因的质粒pPPRE-Luc,及用作转染内参照的带有海肾荧光素酶的质粒phRL-TK,转染24 h后换用含待测样品的培养基,同时设立空白对照(未转染的细胞)、阴性对照(转染的细胞不加样品)和阳性对照[转染的细胞加入匹格列酮(Pioglitazone)],继续培养24 h后用双荧光素酶报告基因检测试剂盒(Promega)检测荧光素酶活性,根据检测到的化学发光强度L值计算激动率PPRE{PPRE=[(L1样品-L1空白)/(L1阴性-L1空白)]/[(L2样品-L2空白)/(L2阴性-L2空白)]×100%,其中:L1为萤火虫荧光素酶的化学发光强度,L2为内参照海肾荧光素酶的化学发光强度;样品检测浓度为10 μg·mL-1,检测时设双复孔,重复两次},数据见表5。
2 结果与讨论
以L-苯甘氨酸为原料,先硝化得中间体1,对其α-氨基进行保护(Boc,Fmoc和Z)得2a~2c; 对2c的羧基进一步苄基化得5;2a~2c的硝基还原得3a~3c。
2.1 合成工艺研究
(1)2a~2c: 以Boc2O为酰化剂,10%K2CO3为碱,在1 2.0 mmol,n(1)∶n(Boc2O)=1.0 ∶1.2条件下于30 ℃反应27.9 h,2a的收率达82.5%;放大实验20倍,最高收率达96.1%。
以Fmoc-OSu为酰化剂,10%K2CO3为碱,在THF/H2O为溶剂,pH=9~10, 1 5.0 mmol,n(1)∶n(Fmoc-OSu)=1.00∶0.98的条件下于20 ℃反应0.7 h,2b收率达88.0%。 放大实验5倍或10倍,收率分别为88.4%和88.8%,且产物色泽很好。
以Z-OSu为酰化剂,10%K2CO3为碱,1 5.0 mmol,n(1)∶n(Z-OSu)=1.0∶1.1,在丙酮/H2O中于30 ℃反应约40 h,收率达97.9%。放大反应10倍,收率不变。
(2) 4: 以Zn/NH4Cl/AcOH为还原剂,TLC跟踪显示还原很完全。但采用萃取法进行后处理,收率只有10%左右。TLC分析发现,4主要留在水相。考虑4易于形成内盐或分子间盐,作者采用调节滤液pH的方法,发现有大量淡黄色沉淀析出,由此简易地实现了产物的分离纯化,结果见表1。
(3)3a: 考虑到Boc基团的稳定性,实验采用水合肼还原[13]2a制备3a,收率低于30%。后选择了弱酸性的Zn/NH4Cl/AcOH还原体系,但还原产物复杂,存在Boc基团脱落的情况;进一步弱化体系的酸性,尝试了中性的Zn/CaCl2还原法,发现还原的最低收率也达到79.4%(表2)。 由表2可见,Zn/CaCl2还原2a制备3a具有反应条件温和、操作简单等优点,具有较好工业价值。
(4)3b: 参照文献[10]的方法将2b和试剂分次加入,发现Zn粉和原料会附着在反应瓶上,反应不均匀,收率最高只有33.3%。对文献[10]方法进行改进,采用所有反应物一次性加入的方式,但收率仍然很低(<23.3%),推测是AcOH使反应物黏在一起导致反应效果不好。 参照文献[12]方法将CaCl2改为NH4Cl而且不加AcOH后,TLC分析发现,除了产物点外还有一个杂质点。我们认为,在没加AcOH的情况下,体系酸度不够,反应不彻底,有部分产物停留在中间体状态。由此认为加酸对该反应是必要的,故尝试在反应约30 min后再添加AcOH的分段投料方法。该方法不仅收率超过84.0%,而且能直接得到纯品。究其原因,是因为此种投料方式,不仅解决了Zn粉和原料附着在瓶壁的关键问题,而且调整了反应体系不同阶段的酸度,保证了硝基的彻底还原。

表1 4的制备条件与结果
a1 4.8 mmol,b1 24.0 mmol,其余反应条件同1.2(3)

表2 3a的制备条件与结果
a2a1.6 mmol,b2a3.2 mmol,c2a10.1 mmol,其余反应条件同1.2(3)

表3 3c的制备条件与结果
a2c1.6 mmol,b2c6.3 mmol,c2c30.0 mmol,d2c26.4 mmol,其余反应条件同1.2(3)
(5)3c:2b按照Zn/NH4Cl/AcOH还原法可以高收率地得到产物3b,但2c同法还原制备3c,收率最高只有45.2%。为了高收率地得到3c,尝试了Zn/CaCl2还原法,结果结果表明,Zn/CaCl2还原法同样适宜于3c的合成。
(6) 6c: Zn/NH4Cl/AcOH和Zn/CaCl2还原法都有较强的还原能力,本文采用Zn/NH4Cl/AcOH体系对5c还原为6c,获得了成功,结果见表4。
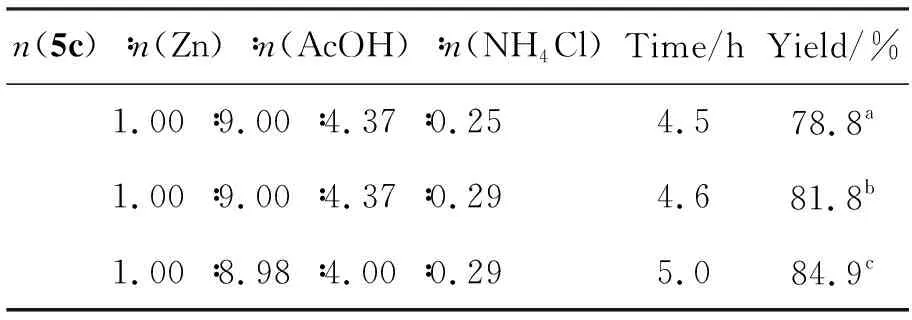
表4 6c的制备条件与结果
a5c1.2 mmol,b5c2.4 mmol,c5c4.8 mmol,其余反应条件同1.2(3)
综上所述,从硝基苯甘氨酸及其衍生物还原目标分子,看起来很简单,但制备中却出现不少困难。起初的收率大多低于45%,且产物很难纯化。通过条件探索,可以稳定地获取高纯度高收率的目标分子,为后续研究奠定了基础。
2.2 生物活性结果
测定了1~6的PPRE数据来判定其抗糖尿病的活性,结果见表5。表5显示,在测定浓度极低的条件下,其中6个化合物依然显示出一定的PPRE激动活性;5个化合物的生物活性达到了10.0%以上(3c和6c的活性分别达到了30.0%和22.8%);2b,2c和3a的活性相近(14.0%左右);5c和3b活性依次降低。分子结构的ChemDraw3D模拟(Chart 1)发现,3c和6c的芳香环处于不同平面内,具有一定的刚性,与吡格列酮有一定的相似性,但是活性差别很大。郭宗儒等[15]指出,配体分子要表现出好的PPARγ活性,必须以U形构象结合在PPAR受体腔内。因此我们推测:3c和6很可能没有以U形构象进入受体腔,因而显示出很低的PPRE激动活性。这些结果提醒我们,要获取活性好的抗糖尿病分子,结构上要尽可能与具有强的PPARγ激动活性的L-酪氨酸衍生物的结构特征一致,亦即必须在3c和6c的苯环氨基引入疏水结构单元。按此思路,作者合成了另外一些L-间氨基苯甘氨酸的苯环氨基修饰物,多种模型上的抗糖尿病活性研究显示,某些分子的活性确实增强(结果另文发表)。

表5 化合物的PPRE活性数据*

Chart 1
3 结论
以L-苯甘氨酸为原料,合成了9个L-苯甘氨酸衍生物,其中5个为新化合物。工艺研究结果表明,该方法具有合成方法简便、成本低廉、收率稳定优良等优点,具有较高的工业应用价值。
致谢:亢为和王宁老师测定IR和NMR,在此深表谢意!
[1] Titulaer G T M, Zhu J, Klunder A J H,etal.Synthesis ofp-hydroxy phenylglycine and some analogues fromp-benzoquinone[J].Org Lett,2000,2(4):473-475.
[2] MENG Y, HU W X, WANG L G.Preparation of chlorophenylglycine as pharmaceutical intermediate[J].Zhe Jiang Chem Ind,2006,37(9):24-26.
[3] 成峰,沈建华,罗小民,等. PPARγ激动剂进展研究[J].中国药物化学杂志,2003,13(2):110-124.
[4] Sassatelli M, Debiton É, Aboab B,etal.Synthesis and antiproliferative activities of indolin-2-one derivatives bearing amino acid moieties[J].Eur J Med Chem,2006,41:709-716.
[5] Lee Y, Marletta M A, Martasek P,etal.Conformationally-restricted arginine analogues as alternative substrates and inhibitors of nitric oxide synthases[J].Bioorg Med Chem,1999,7:1097-1104.
[6] 孔令强,苏小燕,汪林发,等. L-对硝基苯丙氨酸的合成工艺研究[J].西南师范大学学报自然科学版,2009,31(9):102-105.
[7] Rajesh S, Ami E, Kotake T,etal.An expedient synthesis ofNα-protected-L-tetrahydrofuranylglycine and its application in the synthesis of novel substrate based inhibitors of HIV-1 protease[J].Bioorg Med Chem Lett,2002,12:3615-3617.
[8] 许荩,范莉,付成松,等.L-苯丙氨酸与精左氨基物偶联反应的研究[J].西南师范大学学报自然科学版,2009,31(6):184-188.
[9] 宋小礼,范莉,谷光娜,等.N-叔丁氧酰基-O-烷基-L-酪氨酸甲酯的合成工艺研究[J].西南师范大学学报自然科学版,2009,31(7):112-115.
[10] 杨大成,范莉.N-叔丁氧羰基-β-环己基天冬氨酸苄酯的合成研究[J].化学研究与应用,2003,15(4):498-500.
[11] 王乾,秦海芳.Zn粉还原法制备2-氟-5-硝基苯胺[J].化学工业与工程,2008,25(3):271-272.
[12] 胡卫兵,冯驸,刘红霞,等.三氨基芴席夫碱系列化合物的合成及抗菌活性[J].应用化学,2009,26(4):489-491.
[13] Khan F A, Dash J, Sudheer C,etal.Chemoselective reduction of aromatic nitro and azo compounds in ionic liquids using zinc and ammonium salts[J].Tetrahedron Lett,2003,44:7783-7787.
[14] Vass A, Dudás J, Tóth J,etal.Solvent-free reduction of aromatic nitro compounds with alumina-supported hydrazine under microwave irradiation[J].Tetrahedron Lett,2001,42:5347-5349.
[15] 冯君,郭彦伸,陆颖,等.PPAR激动剂的定向设计、虚拟筛选及合成[J].化学学报,2004,62(16):1544-1550.
猜你喜欢
上海计量测试(2022年4期)2022-02-01
云南化工(2021年11期)2022-01-12
世界农药(2019年3期)2019-09-10
福建教育学院学报(2019年5期)2019-07-12
中学生数理化·高二版(2016年3期)2016-12-26
合成化学(2015年10期)2016-01-17
人间(2015年11期)2016-01-09
邵阳学院学报(自然科学版)(2015年2期)2015-06-05
中国洗涤用品工业(2015年7期)2015-02-28
应用化工(2014年9期)2014-08-10
