
cAMP信号分子在缺血后适应心肌保护机制中的作用
2011-11-13 07:57:06邓凤君林焕冰徐江平
中国病理生理杂志 2011年8期
王 茜, 邓凤君, 林焕冰, 徐江平△
(1南方医科大学药学院神经药理课题组,广东广州510515;2益阳医学高等专科学校药理教研室,湖南益阳413000)
再灌注治疗是急性心肌梗死的重要治疗手段。然而再灌注治疗在迅速恢复缺血心肌的血流、挽救大量濒死心肌的同时也导致心肌明显损伤,即“缺血再灌注损伤 (ischemia reperfusion injury,I/R)”。缺血再灌注损伤的发生降低了临床治疗效果。建立减少缺血再灌注损伤的治疗方案是最近30年急性心肌梗死治疗的重要研究方向。有研究发现,在心脏缺血发生之前进行缺血预适应干预可以发挥心脏保护作用,但由于缺血事件不可预知性,该措施则难以应用。近年大量实验研究与临床应用表明,在心肌缺血后、恢复长期血流之前,进行一次或数次短暂重复的缺血/再灌注处理,即缺血后适应(ischemic postconditioning)能提高缺血心肌对之前发生的较长时间缺血的耐受性。与预适应相比,它具有更好的可预测性和临床可控性[1]。但缺血后适应也可能会导致潜在的并发症。因而有学者提出“药物后适应”,即通过使用模拟内源性机制的药物来发挥后适应的保护作用。而应用药物后适应则需要对后适应的机制进行研究。
第二信使环磷酸腺苷(cyclic adenosine monophosphate,cAMP)参与对心脏广泛的细胞功能和形态学过程,包括心肌变力状态、变时性、凋亡和肥大的调节过程。且有研究发现活化腺苷酸环化酶(adenyl cyclase,AC)/cAMP通路的药物能引出缺血心肌的延迟保护作用[2]。但cAMP在后适应中的作用还不是很清楚。因此我们采用离体心脏灌流模型,观察后适应对心脏组织的保护作用,以及AC抑制剂SQ22536和磷酸二酯酶4(phosphodiesterase 4,PDE4)抑制剂洛利普兰(rolipram)对后适应的影响,探讨cAMP在后适应保护心脏中的作用。
材料和方法
1 材料
Sprague-Dawley(SD)大鼠(210-260 g)由广州南方医科大学实验动物中心提供,合格证号为粤检证字第2006-0015号;PCLab生物信号采集处理系统(北京微信斯达科技发展有限公司);肌酸激酶(creatine kinase,CK)和乳酸脱氢酶(lactate dehydrogenase,LDH)测试盒均购自南京建成生物工程研究所,洛利普兰和SQ22536购自Sigma-Aldrich。
2 方法
2.1 实验分组与模型建立 51只大鼠随机分为6组。2.5%戊巴比妥钠 (50 mg/kg)腹腔注射麻醉,舌下静脉注射肝素1 000 IU/kg抗凝。开胸迅速取心脏,置于4℃改良K-H液中,排尽残血后经主动脉将心脏悬挂于Langendorff灌流装置上,用37℃经混合气(95%O2和5%CO2)平衡的K-H缓冲液行主动脉逆行灌流(灌注压为80 cmH2O)。K-H液成分(mmol/L):NaCl 118,KCl 4.7,CaCl21.25,MgSO41.2,NaHCO325,KH2PO41.2,glucose,11.1。实验分组:(1)正常组(control,n=8):K-H液持续灌流120 min;(2)缺血再灌注组(I/R group,n=11):以K-H液平衡15-20 min,37℃停灌45 min,再灌注60 min;(3)后适应组(post,n=10):再灌前给予6个循环的30 s I/30 s R;(4)缺血再灌注后适应+洛利普兰组(post+roli 10μmol/L,n=8):以KH液平衡15 -20 min,改用含10μmol/L洛利普兰的K-H液灌流10 min,停灌45 min,行后适应处理并再灌注60 min;(5)缺血再灌注后适应+SQ22536组(post+SQ22536,n=7):以 K-H液平衡15-20 min,改用含500 mmol/L SQ22536的K-H液灌流10 min,停灌45 min,行后适应处理并再灌注60 min;(6)缺血再灌注+洛利普兰组(I/R+roli,n=7):不行后适应处理,余下过程同缺血再灌注后适应+洛利普兰组。
2.2 左室功能检测 心脏复跳后,将一连接压力换能器的球囊插入左心室,压力换能器与PClab多导生理记录仪相连,在电脑上连续记录左室内压变化,并计算左室发展压(left ventricular developed pressure,LVDP)=最大心室内压-最小心室内压。同时记录冠脉流量(coronary flow,CF)。
2.3 组织病理学检查 再灌60 min后取少许心脏组织4%甲醛固定,石蜡包埋切片,HE染色,光镜下进行形态学检查。
2.4 冠脉流出液酶学检测 分别于停灌前、再灌流60 min时收集冠脉流出液以测定其中LDH和CK含量(按试剂盒说明操作)。
2.5 Total RNA抽提及 real-time PCR 再灌流60 min后取心肌组织,用Trizol试剂提取总RNA,取3μg逆转录合成cDNA。应用Primer 5.0引物设计软件进行引物设计。各基因引物序列如下:β-actin正义链 5'-AGAGGGAAATCGTGCGTGAC -3',反义链5'-CCATACCCAGGAAGGAAGGCT-3';caspase-3正义链 5'-GGAGCAGAGCCATGGGCACG -3',反义链5'-GCACAGACGCCCTGATGGGG -3';bax正义链5'-GGCCCACCAGCTCTGAACAG -3',反义链5'- GCAATCATCCTCTGCAGCTCCATATTA -3';bcl-2正义链5'- TGGAGAGCGTCAACAGGGAGATGTAC -3',反义链 5'- TGGAGAGCGTCAACAGGGAGATGTAC-3'。用Oligo dT18引物逆转录获得总cDNA,实施实时荧光定量PCR反应。以ΔCt表示目的基因与β-actin Ct值的差值、ΔΔCt表示对照组与处理各组 ΔCt的差值,2-ΔΔCt反映目的基因mRNA的相对含量。
3 统计学处理
计量数据以均数±标准差(¯x±s)表示,采用SPSS13.0软件处理,组间显著性检验采用单因素方差分析和LSD检验。
结 果
1 I/R对离体心脏的影响
将离体心脏37℃停灌30、40和45 min,以模拟在体心脏缺血缺氧。结果显示随着停灌时间的延长,LVDP恢复率随之下降,分别由79.61%减至61.45%,54.15%和37.71%。停灌45 min时,CF也显著减少(P<0.05),见图2。冠脉流出液中LDH和CK分别由352.12 U/L和1.90 kU/L增至615.71 kU/L和2.61 kU/L(P<0.05)。并且心肌组织内凋亡调节基因caspase-3表达以及bax/bcl-2比值显著增高,见图5、6。组织病理学检查显示:I/R处理组心肌细胞排列杂乱,横纹模糊不清或消失,细胞肿胀并出现明显空泡样变性、嗜酸性变及凝固性坏死,见图7B。说明该离体心脏灌流模型可以模拟心肌I/R、具有可行性。
2 后适应对离体I/R心脏功能的保护作用
再灌流60 min后将LVDP和CF进行比较,后适应处理能显著增加LVDP(图1)和CF(图2)。停灌前各组冠脉流出液中CK及LDH活性差异无显著。后适应处理能显著降低冠脉流出液中CK及LDH活性(图3和4)。检测再灌流60 min后心肌组织内凋亡相关基因,也得到相似结果,即后适应处理能显著降低心肌组织内caspase-3 mRNA表达(图5)和bax/bcl-2(图6)比值。组织病理学检查可见后适应组部分空泡样变性及嗜酸性变,但细胞损害程度明显低于I/R组(图7)。

Figure 1.Effects of different treatment on LVDP recovery rate.I/R:ischemia/reperfusion;post:postconditioning;roli:rolipram;SQ:SQ22536.¯x±s.n=7-11.*P<0.05 vs control group;#P<0.05 vs I/R group;△P<0.05 vs post group.图1 不同处理对LVDP恢复率的影响

Figure 2.Effects of different treatment on coronary flow(CF).Values were expressed as the percentages of CF measured during equilibrium.¯x±s.n=7-11.*P<0.05 vs control group;#P <0.05 vs I/R group;△P <0.05 vs post group.图2 不同处理对CF的影响

Figure 3.Effects of different treatment on LDH release into coronary artery flow.¯x±s.n=7-11.*P<0.05 vs control group;#P<0.05 vs I/R group;△P <0.05 vs post group.图3 不同处理对心肌缺血再灌时冠脉流出液LDH释放的影响
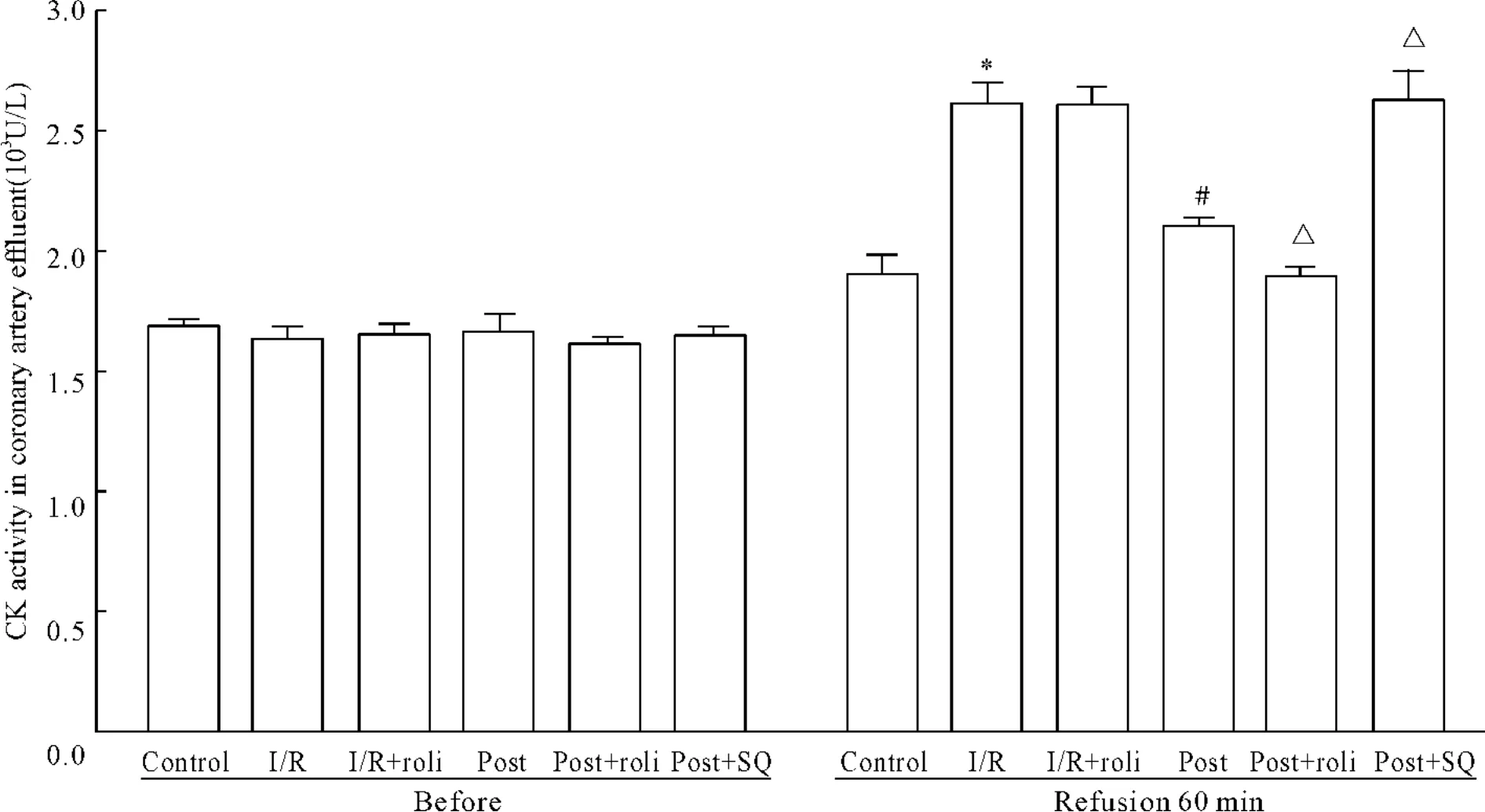
Figure 4.Effects of different treatment on CK release into coronary artery flow.¯x±s.n=7-11.*P<0.05 vs control group;#P<0.05 vs I/R group;△P <0.05 vs post group.图4 不同处理对心肌缺血再灌时冠脉流出液CK释放的影响
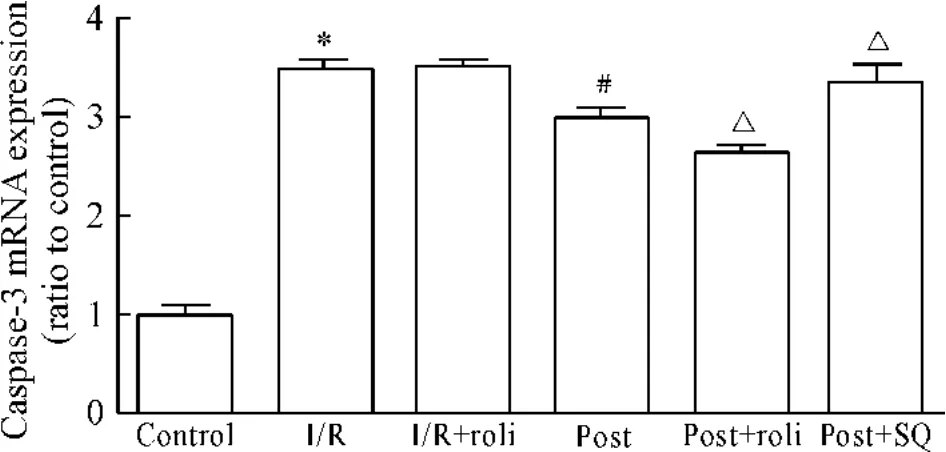
Figure 5.Effects of different treatment on caspase-3 mRNA expression in myocardium.¯x±s.n=6.*P<0.05 vs control group;#P <0.05 vs I/R group;△P <0.05 vs post group.图5 不同处理对心肌缺血再灌时心肌组织caspase-3 mRNA表达的影响
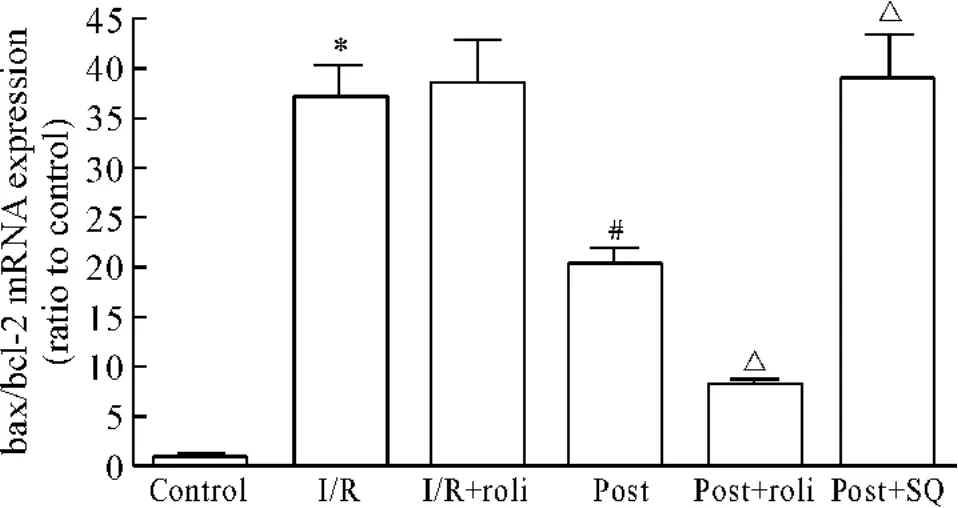
Figure 6.Effects of different treatment on bcl-2/bax ratio in myocardium.¯x±s.n=6.*P<0.05 vs control group;#P <0.05 vs I/R group;△P <0.05 vs post group.图6 不同处理对心肌缺血再灌时心肌组织bcl-2/bax的影响
3 cAMP参与后适应对离体I/R心脏的保护作用
10μmol/L PDE4抑制剂洛利普兰能显著增强后适应对心脏的保护作用,表现为:与后适应组相比,LVDP显著增高(图1)及CF显著增加(图2),冠脉流出液中LDH和CK活性均更为降低,见图3、4。心肌组织内凋亡相关基因检测结果也显示洛利普兰能进一步增强后适应的作用,即caspase-3 mRNA表达减少(图5)、bax/bcl-2(图6)比值降低。而 AC抑制剂SQ22536则能减弱/取消后适应对心脏的保护作用,LVDP显著降低(图1)及CF显著减少(图2),冠脉流出液中LDH和CK活性均增高(图3、4)、心肌组织内caspase-3 mRNA表达增加(图5)、bax/bcl-2(图6)比值上升。并且单用洛利普兰则不能表现出心脏的保护作用,LVDP、CF、酶学指标和凋亡相关基因结果均显示与I/R组无显著差别。组织病理学检查可见后适应与洛利普兰共同处理能减轻心肌组织变性及其它病理改变程度,而后适应与SQ22536共同处理则组织损害程度重于后适应组,见图7。
讨 论
缺血再灌注被认为是多因素参与、非抗原依赖性的炎症反应过程。临床上,心肌I/R常常引起心律失常、心肌顿抑、心肌冬眠、无复流、心肌细胞损伤、LDH及CK释放增加和心功能障碍等病理生理变化[3]。本研究显示,对 Langendorff心脏灌流[4]进行停灌/复流处理,使其出现了LVDP降低和CF减少、心肌LDH和CK释放增加、凋亡/抗凋亡基因表达异常、组织病理学变化,并且凋亡基因mRNA表达增高、抗凋亡基因表达降低。这些结果均与前人一致,说明该离体心脏灌流模型可以用来模拟心肌I/R。

Figure 7.The pathological examination of myocardium in different groups(HE staining,×l00).A:control;B:I/R;C:post;D:post+roli;E:post+SQ;F:I/R+roli.图7 各处理组心脏组织学改变
缺血后适应是一种在心肌再灌注时进行干预的保护措施,2003年Zhao等[5]首次报道了这种干预可导致心脏梗塞面积减少。随后大量研究在其它种属的动物如大鼠[6]、小鼠[7]及家兔[8]也证明了缺血后适应具有保护心脏的能力,甚至在临床实施经皮冠脉介入治疗的病人实行数次短暂的球囊扩张也能够减轻缺血损伤并改善心肌的灌注。目前对于缺血后适应介导心脏保护的机制尚未完全弄清。本研究采用了特异性PDE4抑制剂洛利普兰和AC抑制剂SQ22536两个工具药来观察cAMP在后适应介导心脏保护机制中的作用。结果显示洛利普兰能进一步地增强后适应的保护作用,而SQ22536则抑制了该保护作用。另一方面,洛利普兰在没有进行后适应干预时并未表现出其保护I/R心脏的作用,也说明了并不是由于cAMP自身浓度增高发挥作用的,该结果与前人研究理论一致,即PDE4抑制剂只有在心肌受刺激的状态下影响心肌兴奋性,而单独应用其并不能保护缺血心肌[9]。而后适应这种保护性刺激在cAMP浓度改变时作用增强或减弱,说明cAMP确实参与了后适应对I/R心脏的保护机制。
关于I/R引起的心肌细胞凋亡最初见于1994年Gottlieb等[10]的报道,随后越来越多的动物实验和临床病理标本均证实I/R导致细胞凋亡的产生。也有不同研究发现缺血预适应、缺血后适应均表现出一定的抗心肌细胞凋亡效应[11,12]。细胞凋亡过程的分子调控机制非常复杂,其中bcl-2基因具有抗凋亡作用,而bax基因则可对抗Bcl-2蛋白抗凋亡的作用,细胞中bax和bcl-2 mRNA比值决定了细胞是否发生凋亡。Caspase-3位于Bcl-2和Bax的下游,是凋亡事件的最终效应酶,有研究表明它在心肌I/R损伤中发挥着非常重要的作用。因此本研究中主要对caspase-3 mRNA表达情况和bax/bcl-2比值进行观察,并且再次证实了缺血后适应具有抗心肌细胞凋亡并改善心功能的作用。而且采用洛利普兰能增强后适应的抗凋亡作用,而SQ22536能抑制该作用,因此认为cAMP信号分子参与了后适应调节心肌细胞凋亡信号的机制,即后适应通过增加该信号组分抗凋亡、保护心肌。
[1] Hausenloy DJ,Yellon DM.Preconditioning and postconditioning:underlying mechanisms and clinical application[J].Atherosclerosis,2009,204(2):334 -341.
[2] Szekeres L.Drug-induced delayed cardiac protection against the effects of myocardial ischemia[J].Pharmacol Ther,2005,108(3):269 -280.
[3] Skyschally A,Schulz R,Heusch G.Pathophysiology of myocardial infarction:protection by ischemic pre- and postconditioning[J].Herz,2008,33(2):88 - 100.
[4] Vidavalur R,Swarnakar S,Thirunavukkarasu M,et al.Ex vivo and in vivo approaches to study mechanisms of cardioprotection targeting ischemia/reperfusion(I/R)injury:useful techniques for cardiovascular drug discovery[J].Curr Drug Discov Technol,2008,5(4):269 -278.
[5] Zhao ZQ,Corvera JS,Halkos ME,et al.Inhibition of myocardial injury by ischemic postconditioning during reperfusion: comparison with ischemic preconditioning[J].Am J Physiol Heart Circ Physiol,2003,285(2):H579-H588.
[6] Pinheiro BB,Fiorelli AI,Gomes OM,et al.Cardiac effects of postconditioning depend critically on the duration of reperfusion and reocclusion episodes[J].Heart Surg Forum,2010,13(1):E52-E56.
[7] Kaljusto ML,Rutkovsky A,Stenslokken KO,et al.Postconditioning in mouse hearts is inhibited by blocking the reverse mode of the sodium - calcium exchanger[J].Interact Cardiovasc Thorac Surg,2010,10(5):743 -748.
[8] Donato M,D'Annunzio V,Buchholz B,et al.Role of matrix metalloproteinase-2 in the cardioprotective effect of ischaemic postconditioning[J].Exp Physiol,2010,95(2):274-281.
[9] Rao YJ,Xi L.Pivotal effects of phosphodiesterase inhibitors on myocyte contractility and viability in normal and ischemic hearts[J].Acta Pharmacol Sin,2009,30(1):1-24.
[10] Gottlieb RA,Burleson KO,Kloner RA,et al.Reperfusion injury induces apoptosis in rabbit cardiomyocytes[J].JClin Invest,1994,94(4):1621 -1628.
[11] Sun H,Guo T,Liu L,et al.Ischemic postconditioning inhibits apoptosis after acute myocardial infarction in pigs[J].Heart Surg Forum,2010,13(5):E305-E310.
[12] Cheng Y,Zhu P,Yang J,et al.Ischaemic preconditioning-regulated miR-21 protects heart against ischaemia/reperfusion injury via anti-apoptosis through its target PDCD4[J].Cardiovasc Res,2010,87(3):431 -439.
猜你喜欢
中华养生保健(2020年7期)2020-11-16 01:14:24
山东林业科技(2019年2期)2019-06-03 10:10:54
现代园艺(2017年11期)2017-06-28 11:32:46
奥秘(2016年10期)2016-12-17 13:13:11
实用临床医学(2016年8期)2016-06-07 01:28:18
中国卫生标准管理(2015年5期)2016-01-14 05:16:56
中国卫生标准管理(2015年4期)2016-01-14 05:16:48
中国卫生标准管理(2015年3期)2016-01-14 03:41:51
西藏科技(2015年9期)2015-09-26 12:15:31
中国当代医药(2015年22期)2015-03-01 02:05:28
