
金属卟啉类化合物特性及光催化机理与应用研究
2011-10-22 07:24:50罗光富曹婷婷方艳芬黄应平
三峡大学学报(自然科学版) 2011年5期
王 攀 罗光富 曹婷婷 饶 志 方艳芬 黄应平
(三峡大学 三峡库区生态环境教育部工程研究中心,湖北 宜昌 443002)
卟啉及其金属卟啉类化合物应用十分广泛,包括有金属离子的检测[1]、光催化动力学疗法[2]、太阳能的光电转化[3]、液晶材料的制备[4]、选择性催化氧化[5]、光催化环氧化[6]和光催化降解有毒有机污染物等[7].
近年来,卟啉及金属卟啉类化合物在光催化处理有毒有机污染物方面倍受研究者的关注,然而对其光催化作用机理还需要进行深入的研究和探讨.本文从卟啉及金属卟啉的基本性质出发,对卟啉的光电化学性质作了总结,并对其在光催化方面的应用等进行了归纳,重点综述了其光催化氧化作用机理.
1 卟啉类化合物分子结构特性与化学合成
卟啉类化合物是一类中心由20个C和4个N形成的具有一个24个中心26个电子的大π键,并且所有大环原子处于同一平面上的大共轭杂环类芳香性化合物,其中C和N均为sp2杂化,C上P轨道的一个单电子和N上P轨道的孤对电子参与共轭.卟啉和类卟啉化合物的共轭能约为1670~2500kJ/mol,具有较为稳定共轭结构,而中心环16π环18π电子体系对体系的稳定能贡献最大.由于共轭大环的存在,这类化合物在380~420nm之间出现非常强的吸收带,一般具有很深的颜色.卟啉主要吸收带通常称为Soret带(亦称为B带)和Q带,其中B带是卟啉环的a1u(π)-eg(π*)允许跃迁,为强吸收,其吸光系数均为10-4级,而Q带为弱吸收带,它们是卟啉环的a2u(π)-eg(π*)准允许跃迁.中性卟啉的 Q带通常含有4个峰(见图1所示).

图1 卟啉分子的Q带和B带吸收光谱
卟啉因其吡咯环上的-NH键的存在而具有一定的弱碱性.作为弱碱,其pKa1≈7,pKa2≈4,它们可以被质子化形成双阳离子型卟啉.卟啉和它们的金属配合物均可被亲电试剂取代,例如在meso-和吡咯的β位上发生氘代、硝化和Vilsmeier酰化等取代反应,形成各种各样的卟啉及金属卟啉.卟啉类化合物经硼氢化钠、Na/Hg或催化加氢可以得到还原卟啉类化合物.
卟啉化合物是用吡咯或者取代吡咯与各种醛通过缩合反应制得,在合成卟啉过程中,反应条件及方式对卟啉的产率有较大的影响.已有众多经典的合成方法,包括 Alder-Longo法[8]、Lindsey法[9]2+2[10]合成法、和3+1[11]合成法等.这些合成方法各有优缺点,如Alder-Longo法,其操作简单,实验条件不是很苛刻,易于合成无取代及非水溶性取代卟啉,且反应产率较高,但是反应温度较高,其不能选用对酸敏感的醛类作为反应物,同时酸会使吡咯发生聚合,产生大量焦油状的副产物,也给分离纯化带来了一定的困难.Lindsey法是基于还原卟啉的合成,然后再氧化生成卟啉,此法能够克服酸对反应体系的影响,反应的产率较高且易分离纯化,然而其反应体系中原料浓度(一般10-2M)较低,不利于大量合成.2+2和3+1合成法主要应用于不对称卟啉的合成,其合成活性较高,常在常温下进行,反应的副产物较少,是合成卟啉方法中产率最高的方法之一.
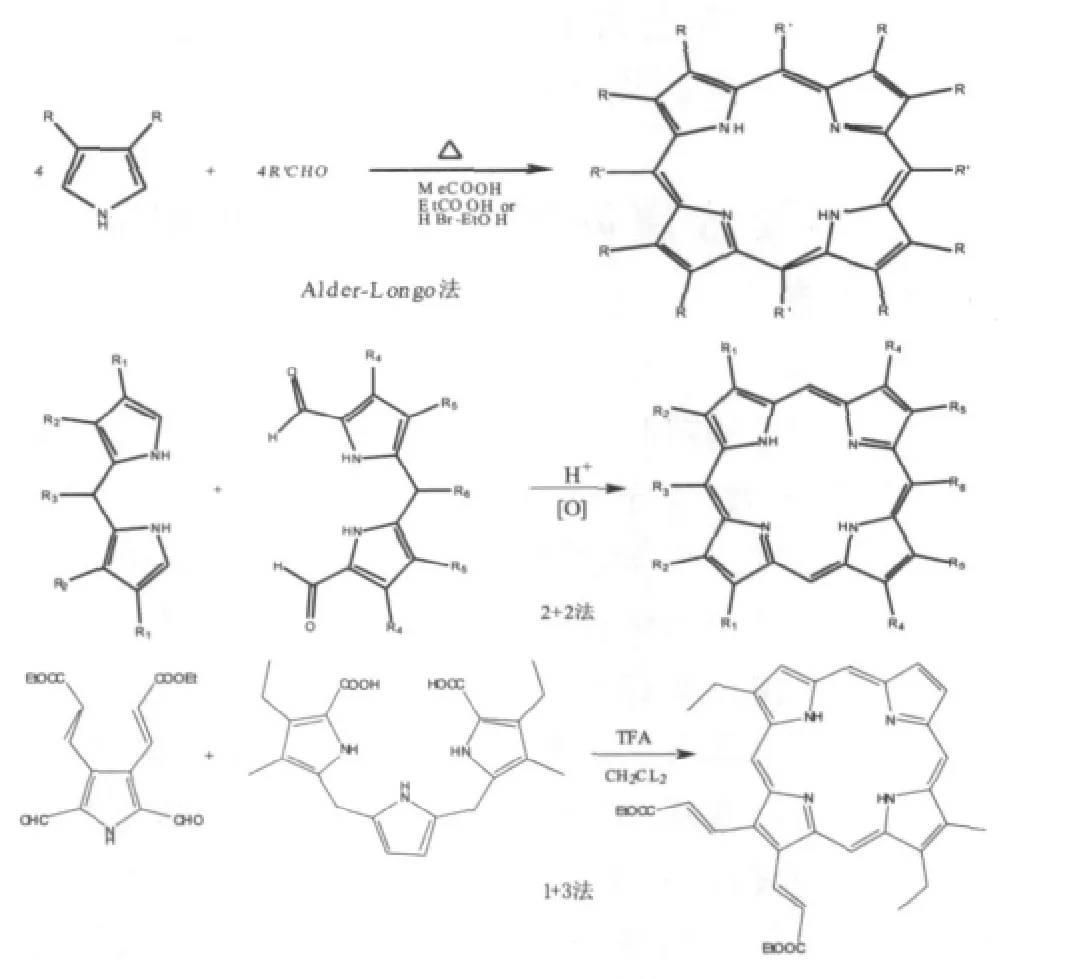
图2 卟啉的合成方法
2 金属卟啉类化合物特性
金属离子进入卟啉环内以后形成的金属配合物称为金属卟啉,对称性较卟啉配体强,吸收峰数目减少.金属卟啉一般为D4h对称,卟啉配体则为D2h对称.卟啉可以与二价金属离子如Co(Ⅱ),Ni(Ⅱ),Cu(Ⅱ)形成不带电的四配位金属卟啉络合物,其中Ni(Ⅱ),Cu(Ⅱ)的卟啉络合物对另外的配体亲和力低;而 Mg(Ⅱ),Cd(Ⅱ)和Zn(Ⅱ)等二价金属离子容易与其他配体继续配位形成五配位络合物;Fe(Ⅱ),Co(Ⅱ),Mn(Ⅱ)能形成变形的八面体络合物[12].卟啉与金属形成配合物的难易程度不同,一般与金属离子的半径有较大关系,如离子半径较大的Hg、Pb及Cd不能进入卟啉配合,只能在卟啉分子的上或者下面反应,形成“坐顶络合物”,这个配合物能使卟啉核变性,易于与其他金属离子配合生成金属卟啉[13].
高价金属卟啉属于金属卟啉配合物,然而中心离子的价态要比一般状态下的金属离子的价态高1到2价,因金属离子的价态升高,其比低价的金属卟啉具有更优异的氧化还原性质,同时与金属中心配位的轴向配体数目也相应的增多,在一定程度上会影响金属催化特性.在活化H2O2及O2过程中,金属离子通常在其轴向上与O结合形成双键,又被称高价卟啉金属氧络合物.高价卟啉金属氧络合物常用于端基的氧化及选择性环氧化方面,如在氯化血红素[14]及辣根过氧化物酶[15]模拟血红素选择性催化氧化烷烃及烯烃的反应体系中,催化剂的本质就是高价卟啉金属氧络合物.在细胞色素P450的催化环氧化过程中的催化剂也属于高价铁物种[16].
3 卟啉及金属卟啉类化合物光电及光催化性质
3.1 光致电子转移
所谓光致电子转移(Photoinduced Electron Transfer PET)[17],即受光激发的物质与未受激发的物质之间的电子的传递,和受光激后的物质将产生的电子由一个位点转移至另一位点的电子的传递.
卟啉由于具有流动性较强的大π共轭结构,作为一个有色染料基团,它在光照的条件下通常都能发生光致电子的转移.在光致电子转移的体系中,卟啉配体常作为电子的供体,在受光激发后,能将光激发后产生的光电子转移至电子受体.卟啉的光致电子转移通常发生在共价结合的体系中,如Baskaran等[18]研究了作为电子供体间位取代的卟啉与作为电子受体的碳纳米管结合后的光致电子转移(图3).研究发现,在550nm激发光照射下,卟啉与碳纳米管共价结合后,在650nm和700nm处的荧光发射淬灭效率达95%~100%.在非共价结合(如:氢键、芳香π堆积、疏水作用等)的超分子自组装体系中,卟啉组装体也能发生光生电子转移的现象.在非共价的光生电子转移的过程中,氢键可以作为电子传递的界面(图3),如Derege研究Zn卟啉和Fe卟啉通过氢键组成体系中的电子传递特性发现:Zn卟啉作为电子的供体,而Fe卟啉作为电子的受体,其间的电子是通过苯甲酸取代基上两个羧基形成的分子间氢键传递的[19].光致电子转移能够有效的降低光致发光效率,提高光能向化学能的转化效率,这样有利于能量的传递.如Shan等将卟啉负载于纳米Pt上制成的催化剂能将光激发产生的电子转移至金属核上,提高了催化剂光催化还原水制氢,有效的将光能转变为化学能,反应过程中伴随着光生电子的转移,经过光电转移后的卟啉中产生了具有氧化活性的类似空穴的物种V+,需要在体系中加入EDTA来有效防止自身的氧化,说明光致电子转移赋予了卟啉催化剂光氧化能力[20].

图3 共价和氢键电子转移
卟啉与金属离子配位生成的金属卟啉因配体的存在具有一定的光致电子转移的特性.一般情况下,金属卟啉中的金属中心具有较高的氧化态,在光电转移过程中常作为电子受体,而卟啉配体则作为光致电子的供体.在光照条件下,卟啉配体将光致电子转移至金属中心,致使光致电荷分离,产生了类似半导体的具有催化氧化性和还原性的电子-空穴对,赋予了金属卟啉的光催化性质.
3.2 卟啉的光致激发态能量转移
光致电子传递能够促进光致激发态能量的转移,即光致激发态能量转移的过程中可以伴随着电子的传递(如图4),光致电子转移的结果往往导致光致电荷的分离,从而使电子受体多电子,而电子供体少电子.激发态能量转移最终是将激发态物质的激发态能量转移给未激发的底物,使底物变为激发态,自身则还原为基态,转移前后激发物与底物各自并未发生电子的得失.物质之间能发生能量转移的前提条件是激发态物质发射光谱的能量范围要与底物的吸收光谱的能量范围发生重叠(如图5).

图4 电子传递能量转移
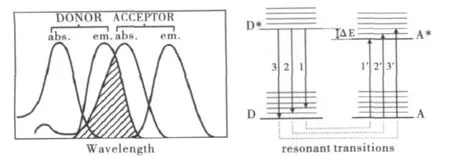
图5 供体和受体间的能量要求
能量转移可分为两大类,即辐射转移和无辐射转移.能量转移可以产生于不同的作用机理,其中包括Förster机理和 Dexter机理[21].所谓 Förster机理即能量的转移受自旋规则的限制,一般只存在单线态-单线态(1D*+1A→1D+1A*)和单线态-三线态(1D*+3A→1D+3A*)的能量转移.而Dexter理论则是基于分子间电子云重叠作用的电子交换转移.同Förster机理相比,Dexter机理只需要给体-受体分子对的电子云有效的交叠,不论单线态-单态的能量转移,还是三线态-三线态的能量转移均是允许的,即D*+A→D+A*.卟啉的基态属于单重态(0S),受光激发后优先生成激发单重态(1S*),然后可以转化为激发三重态(3S*),在发生能量的转移过程中可以利用激发单线态活化单线态物质形成激发单线态,或者是活化三线态物质成激发三线态(如Förster机理所述).另一方面,激发态卟啉转变为激发三线态后能够将基态三线态物质活化为激发单线态和更高的激发三线态(如Dexter机理所述),而基态的3O2为三线态,这样就赋予了卟啉光敏化能量转移活化分子3O2产生具备更高氧化活性的1O2的性质.
3.3 金属卟啉氧化物种的种类、产生及性质
金属卟啉具有光致电子传递和光激发能量转移的性质,这些光电性质都有助于它在光催化方面的应用.然而金属卟啉除了具有上述光电性质外,还具有高价金属卟啉氧化物种这一特殊的化学状态,这一性质也被作为金属卟啉催化机理的一个方面,引起了研究者的关注.金属卟啉氧化物种类较多,如高价锰氧卟啉、高价铬氧卟啉[21]、高价钌氧卟啉以及 Mo、Nb、Ti、V等高价金属氧卟啉[22],只要是金属卟啉的金属中心具有变价,其均能形成金属卟啉氧化物种,因它们在反应过程中通常以中间体的形式存在,又可称其为变价金属卟啉类化合物.
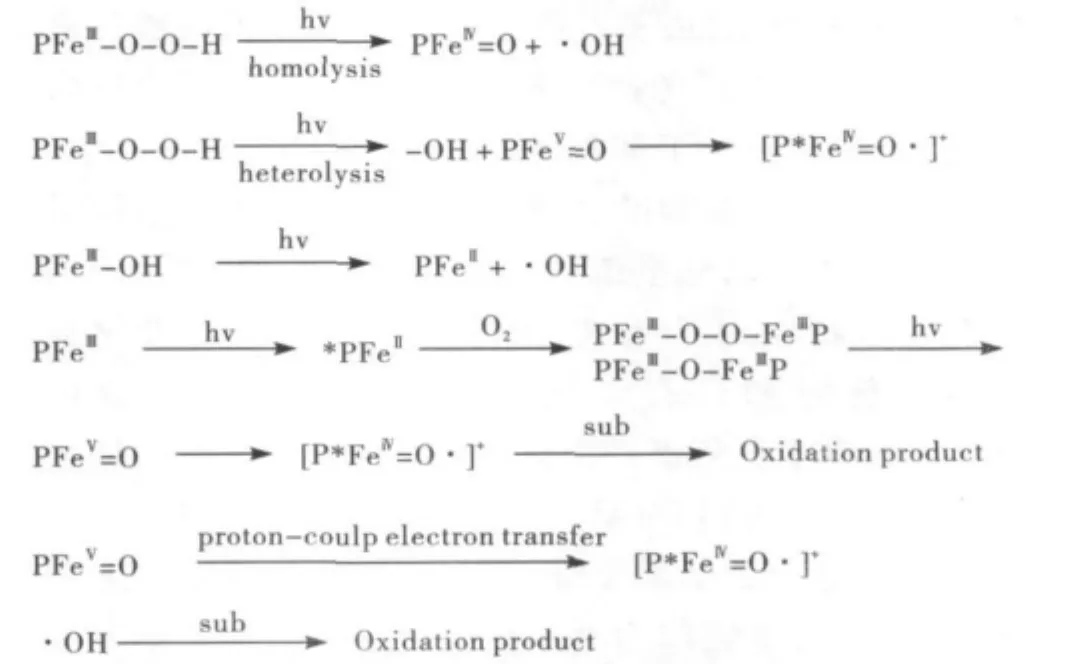
变价金属卟啉氧化物种的产生在初期常常伴随着氧化剂的氧化,以高价铁氧卟啉化合物为例,其产生通常由铁(Ⅲ)卟啉与端基氧化物反应制得,如:间氯苯甲酸、亚碘酰苯和双氧水等[23].在选择性氧化反应中以中间氧化产物的形式存在而体现其催化特性.在变价卟啉氧化物种催化氧化的过程中,因金属离子与氧之间键的断裂方式的不同,产生的中间氧化物种也不同,通常情况下,异裂产生氧化物种FeV=O.因在反应的过程中常伴随着电荷的分离及自由基信号的产生,金属卟啉可被称为高价金属卟啉π阳离子自由基,如:铁(Ⅳ)氧卟啉π阳离子自由基([(Porp)+.FeIV=O]+),而均裂则产生FeⅣ=O,其可以通过质子配对电子转移的方式转变为 ([(Porp)+.FeIV=O]+).在细胞色素P450中,低自旋的过氧羟基铁卟啉通过异裂的方式产生一个FeV=O物种,这个物种可以更准确的用[FeIV=O(*Por)]+来表示,其自由基阳离子的产生反映在配体的电子自旋离域性上面.高价金属卟啉π阳离子自由基是一个亲电物种,这样有利于其与烯烃等物质的接触来实现其选择性催化氧化[24].
变价金属卟啉氧化物种往往出现在酶催化体系中,酶催化剂通常为Fe、Cu的变价金属卟啉化合物,在生物体中通常与氧结合,扮演着运输和活化分子氧的重要角色.如属于血红素酶的辣根过氧化物酶,其既能活化过氧化氢,也能活化分子氧,除了具有过氧化物酶的特点外,也能催化氧化某些底物.变价金属卟啉的催化氧化的机理包含两种,一种是自由基的反应,而另外一种则是氧合过氧化物酶的机理.氧合过氧化物酶在很多方面与氧合肌红蛋白相似,它们都含有一个与组氨酸结合的正铁血红素,同时氧分子作为它们的第五或者第六配体.然而氧合过氧化物酶能高度的活化分子氧,而氧合肌红蛋白则不能活化分子氧,这是因轴向配体的不同使分子氧O-O键的强弱不同导致.Atkinson等利用共振拉曼光谱研究了辣根氧合过氧化物酶和氧合肌红蛋白之间的性质差异,研究发现:含有卟啉环的辣根氧合过氧化物酶的环有轻微的扩展,其Fe中心更接近于卟啉平面,且其较氧合肌红蛋白有较高的Fedx-Oπ*反键轨道,其Fe-O键的拉曼光谱分别为570和562cm-1.这是由于氧合过氧化物酶中的Fe-His键提高了Fe3dx轨道能量,使其更接近于O的π*轨道,形成了更高的Fedx-Oπ*反键轨道的缘故,这样就减弱了O-O键,从而在过氧化物反应体系中作为一个电子受体来活化分子氧参与氧化反应[25].同时,不同价态的高价铁物种的氧化性随着轴向配体的种类、卟啉中心离子的电性及反应的底物的不同而有所不同.如Kang等研究了不同对位取代的吡啶氧作为轴向配体对高价金属卟啉π阳离子自由基的氧化反应活性的影响,发现不同取代的轴向配体的价铁物种的氧化性不同,其氧化活性随着轴向配体的拉电子效应的增强而增强,其氧化活性顺序为1-OCH3>1-CH3>1-H>1-Cl[26].这是因为拉电子轴向配体及阴离子配体能加强Fe-H的键强度,提高了其夺氢活性,同时减弱Fe=O双键的强度,有利于其键的断裂及氧的转移来实现催化氧化.由此可知金属卟啉在一定程度上能活化分子氧,并可通过金属离子及配体的选择来调节其催化特性,具有光催化的潜质.
4 卟啉光催化机理
4.1 卟啉敏化光致电子转移光催化
X.Q等用碘化氨基卟啉(TAPPI)和磺基苯基Co卟啉(TPPSCo)与一维的ZnO复合形成的异相光催化剂,在可见光下活化分子氧光催化降解了RhB,提高了ZnO可见光催化活性,并初步描述了其催化氧化机理[27].最具有典型代表的是卟啉敏化TiO2光催化降解,蔡金华等制备的5-(对-烯丙氧基)苯基-10,15,20-三对氯苯基卟啉(APTCPP)敏化的 TiO2复合微球APTCPP-MPSTiO2有效提高了TiO2对α-松油烯的光催化氧化,催化氧化产物主要是土荆芥油素[28].在光催化氧化过程中,卟啉作为有色染料,将受光激发后产生的电子转移至半导体ZnO或者TiO2的价带,使产生的电子与卟啉配体发生了分离,避免了其光生电子与空穴的复合,有利于价带电子还原分子氧O2产生·O2-、·OH等氧化物种,实现对底物的选择性氧化及降解,光催化氧化机理如图6所示.
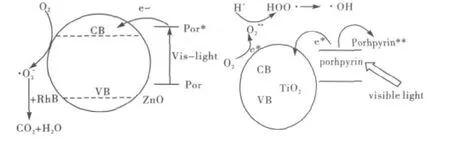
图6 光电子转移及卟啉敏化ZnO和TiO2
作者课题组利用β-CD-Hemin(CDH)光催化降解RhB和二氯酚(DCP),发现其在可见光、H2O2及中性条件下能够很好的氧化RhB及DCP,其矿化率分别可达72%和85%[29],拓宽了Fenton体系的pH应用范围,提高其实际应用性,并具有较高的催化稳定性.在降解过程中,金属卟啉先与H2O2反应形成HOOFeⅢ-L,在光照和β-CD辅助条件下,通过电子由金属到配体的电荷转移(MLCT)导致O=FeIV-L和·OH的产生,由于·OH较高价铁物种具有更高的氧化活性而对有机底物具有较高的氧化矿化效果.说明电子转移存在于金属卟啉配合物类Fenton光催化氧化降解有毒有机污染物体系之间.其机理如下:

Maldotti等在表面活性剂的作用下形成的[Fe(III)(TDCPP)]微乳异相光催化体系在可见光及分子氧的条件下,能将环己烯和环辛烯氧化生成环氧化物、酮和醇等氧化产物.在氧化过程中,[Fe(III)(TDCPP)]在可见光照下发生配体到金属Fe(III)中心的光致电子转移(LMCT),生成[Fe(II)(TDCPP)],使其在轴向上与O2结合后生成铁氧端基自由基,并在烯丙基位置上发生自由基亲电加成反应,生成过氧产物[30],此过氧产物经过异裂和均裂的方式生成酮类物质和醇类物质,其卟啉端在异裂过程中产生了高价Fe氧络合物,参与催化环氧化反应,成功实现了卟啉对分子氧的活化和转移.其机理如图7所示.
S D.G等利用苯基卟啉及其Cu、Ag和Sn的金属卟啉在太阳光及不同的pH条件下光催化降解甲基橙,发现在氧气饱和的溶液中,金属卟啉能够有效降解甲基橙,测定其催化降解的活性能力大小为TPP<CuTPP<AgTPP<SnTPP.并推测机理与半导体光催化机理中的空穴与电子类似[31],其中也涉及到光致电荷的分离.
综上表明,在卟啉类化合物的光催化降解过程中,常常伴随着光致电子转移及分离,产生的分离态电子或空穴以实现卟啉类化合物的光催化活性,是卟啉类化合物光催化机理的一个方面.
4.2 卟啉敏化能量转移光催化
H.J等采用四磺基卟啉及Cu、Fe卟啉在未加任何氧化剂的情况下就能催化氧化降解TNT,生成三硝基苯甲酸和三硝基苯[32],虽然文中未能对其光催化机理作较为深入的研究,但可以初步推测其催化氧化过程可能涉及到光致能量转移活化分子氧历程.
J.H 等将5-(4-烯丙氧基)苯基-10,15,20-三(2,6-二氯苯基)卟啉用3-巯基丙基三甲氧基硅烷修饰后负载于纳米SiO2球上用于可见光光催化降解1,5-二羟基萘,发现其能很好的催化氧化1,5-二羟基萘,并且其催化降解速率与氧气的浓度呈正比,说明此卟啉修饰的纳米二氧化硅催化剂能活化分子氧催化氧化无色小分子物质[33].众所周知SiO2为惰性载体,其导带不能为电子传递所用,故此催化剂不能产生光致电荷分离,而文中卟啉具有活化分子氧的能力,表明卟啉可以不通过光生电子的传递来活化分子氧来产生氧化物种.
S D,G等将苯基卟啉及其金属卟啉(银、铜和锡)应用于异相光催化降解甲基橙,其催化降解机理涉及到敏化活化分子氧的氧化机理[31].W.K,J P等利用可溶性及非水溶性Sn卟啉负载SiO2进行了异相光催化降解4-氯苯酚和AO7,其氧化机理为活化分子氧机理[34].C.J,P.M 在研究水溶性卟啉光敏化降解二氯苯酚的机理过程中,采用激光作为敏化光源,同时运用对红外线敏感的光电倍增管测定了单线态氧在1270nm处淬灭时发射光谱,并由此计出TDCPPS、ZnTDCPPS和SnTDCPPS的单线态氧量子效率,分别为0.83%、0.55%和0.61%,更加确切地证明了单线态氧的存在[35].
综上所述,在卟啉类化合物的光催化降解过程中,除了光致电子转移及分离产生的分离态的电子或空穴外,激发态卟啉类化合物能量转移活化分子氧及底物也能实现卟啉类化合物的光催化氧化,是卟啉类化合物光催化机理的一个方面.其机理可概括如图8所示.

图8 卟啉敏化能量转移活化分子氧和底物
4.3 变价金属卟啉光催化
C.C J在研究中报道了锑卟啉在光照条件下具有活化分子氧的功能,在其光催化活化分子氧历程中经历了双电子或者是四电子还原氧分子的过程,其锑卟啉活化分子氧产生双氧水的历程可简述为:

K.C等将Fe卟啉负载于纳米SiO2上用于五氯酚的氧化降解,发现在光催化条件下,催化剂能实现对五氯酚的高效氧化转化,并在实验过程中采用EPR和DR-UV-Vis光谱技术验证了高价Fe氧卟啉盐离子自由基的存在[37].
Manhdi等利用卟啉敏化剂在光照条件下选择性环氧化环庚烯,其反应过程中伴随着变价金属卟啉物种的产生.
综上所述,卟啉在体现其催化氧化过程中常伴随着高价金属卟啉物种的产生,作为中间氧化物种的金属卟啉物种具有一定的选择性催化氧化及活化分子氧等氧化剂的能力,是描述其光催化过程不可缺少的一个环节,其中具有典型代表的高价金属卟啉的是高价铁卟啉和高价锰卟啉[38],其机理可概述如下:
卟啉类化合物的光催化过程较为复杂,其光催化氧化机理也较为多样,各种催化机理之间存在相互的联系,不能为单一的催化机理所能概括.另外,卟啉类化合物中的变价金属卟啉具有更加广阔的探讨空间,其催化活性往往因卟啉配体中取代基电性的不同及金属离子的不同而使氧化能力的大小不同.另外,卟啉的功能多样性可以通过对其基本电子结构的调节来实现,位于中心离子上的电性和轴向配体在卟啉类化合物光催化性质方面起着至关重要的作用,也是影响高价金属卟啉光催化活性的一个主要因素.
5 金属卟啉异相光催化
Konstantinos的异相光催化体系具有比均相的Fe卟啉更高的催化氧化五氯酚转化的效率,且催化剂具有较高的循环利用性[37].同时不同的载体负载对卟啉负载敏化催化剂有较大的影响,这就要求考虑卟啉负载后其与载体连接的稳定性、连接后的活性等因素[35].另外进行载体负载后的光催化机理也会发生相应的改变.
Giuseppe等在文中将四丁基苯基卟啉和其金属卟啉负载于聚晶TiO2上,并将其应用在光催化降解4-硝基酚中,发现负载后的催化剂的催化活性有了较大提高是因为卟啉负载使其光生空穴离域化,从而有了较长的生存时间,更有利于其对底物的光催化氧化[39],与负载前的TiO2自身半导体光催化和卟啉自身的染料敏化机理相比,催化机理发生了改变.
卟啉在光催化的过程中容易产生高活性的氧化物种,这种高氧化活性物种对卟啉自身的稳定性有很大的影响,同时由于卟啉间存在的大π共轭结构及氢键,容易导致卟啉在分子之间发生积聚,阻碍了底物与催化剂位点的接触,从而影响其光催化活性[40].另一方面,由于卟啉存在上述的作用,其易在分子之间发生能量的转移,降低了其活化分子氧的效率,进而影响其光催化活性.将卟啉进行负载一方面能够提高其稳定性,提高其光催化效率,同时也实现了卟啉催化剂的再生,有利于催化剂的循环使用.
如今,较为新型的金属卟啉异相超分子化合物异相光催化体系已见报道[41],其产生与氢键、分子间引力和π堆积作用[42]有关.其中氢键是超分子卟啉体系得以稳定和具有光催化活性的的一个重要因素[43],虽然单个氢键的键能较小,而多个氢键的能量较大,常可以达到50kJ·mol-1的数量级,相当于最弱的共价键或者是配位键,有利于体系的稳定和电子的传导.并且氢键作为电子转移的媒介比长的饱和碳碳键具有更高的转化效率,有利于其电子转移光催化氧化.
6 卟啉的光催化应用
卟啉的光催化应用较为广泛.在选择性氧化方面,利用卟啉类化合物能达到使环烯烃选择性环氧化的目的,实现其拟酶催化特性.如:C J.L等将钌卟啉负载于多孔的MCM-41上,以2,6-二氯氧化吡啶作为端基氧化剂,能高效的将芳烃及烷烃环氧化生成环氧化物[43].
卟啉在处理环境污染物种也有较为广泛的应用,其中包括染料敏化半导体异相光催化降解有毒有机污染物,其能有效的氧化降解有色有机大分子和无色有毒小分子,如:H.H等将卟啉与铁卟啉负载于TiO2用于罗丹明B的降解,发现卟啉负载的TiO2能氧化降解51.19%的罗丹明B[44].M Y.C等将卟啉负载的 TiO2用于光催化降解2,4-二氯苯酚(2,4-DCP),发现当DCP的浓度为10ppm时,其氧化率能达到81%,当浓度为100ppm时,其氧化氯能达到42%,由此表明卟啉负载的TiO2能氧化矿化有毒小分子物质[45].
在光催化动力学疗法中,应用卟啉敏化活化分子氧的特性,来实现有有毒组织的氧化,从而达到治疗癌症等疑难病的目的.如:S.G等将血卟啉衍生物用于恶性肿瘤的光动力学疗法,发现其能在短时间内就能使肿瘤细胞坏死[46].
7 展 望
由于卟啉及其金属卟啉具有上述良好的光电化学性质,在太阳能的转化中实现更高的能量转化效率,同时基于其特殊的光催化氧化和可见光响应的特性,其在处理环境有毒有机污染物上面有着广泛的应用.最后,卟啉之间存在大π共轭结构及分子间相互作用能够使其自组装成超分子体系,这种超分子体系能够通过分子间作用实现光电子的传递和分离,将有利于其光催化降解有毒有机污染物.卟啉催化剂的研究还处在较新的阶段,深入的研究将会揭示更多其特殊的光电化学效应及应用.
[1]Yang R,Kean L,Wang K,et al.Porphyrin assembly onβ-cyclodextrin for selective sensing and detection of a zinc ion based on the dual emission fluorescence ratio[J].Analytical chemistry,2003,75(3):612-621.
[2]Ethirajan M,Chen Y,Joshi P,et al.The role of porphyrin chemistry in tumor imaging and photodynamic therapy[J].Chem.Soc.Rev,2011,40,340-362.
[3]Basham J,Mor G K,Grimes C A.Forster Resonance Energy Transfer in Dye-Sensitized Solar Cells[J].ACS nano,4(3):1253-1258.
[4]Sun E J,Sun Z Y,Yuan M,et al.The synthesis and properties of meso-tetra(4-alkylamidophenyl)porphyrin liquid crystals and their Zn complexes[J].Dyes and Pigments,2009,81(2):124-130.
[5]Choi K S,Chiu P F,Chan K S.Selective Activation of Benzylic Carbon-Hydrogen Bonds of Toluenes with Rhodium (III)Porphyrin Methyl:Scope and Mechanism[J].Organometallics,29(3):624-629.
[6]Wu J,Liu C,Jiang Y,et al.Synthesis of chiral epichlorohydrin by chloroperoxidase-catalyzed epoxidation of 3-chloropropene in the presence of an ionic liquid as co-solvent[J].Catalysis Communications,11(8):727-731.
[7]Granados O G,P E A,Ortega F M,et al.Degradation of atrazine using metalloporphyrins supported on TiO2under visible light irradiation[J].Applied Catalysis B:Environmental,2009,89(3-4):448-454.
[8]Adler A D,Longo F R,Finarelli J D,et al.A Simplified Synthesis for meso-Tetraphenylporphine [J].J.Org.Chem.1967,32(2):476-476.
[9]Littler B J,Ciringh Y,Lindsey J S.Investigation of Conditions Giving Minimal Scrambling in the Synthesis of trans-Porphyrins from Dipyrromethanes and Aldehydes[J].J.Org.Chem.,1999,64(8):2864-2872.
[10]Arsenault G P,Bullock E,MacDonald S F.Pyrromethanes and Porphyrins Therefrom[J].J.Am.Chem.Soc.1960,82(16):4384-4389.
[11]Chmielewski P J,Latos-Grazyński L,Rachlewicz K.5,10,15,20-Tetraphenyl sapphyrin-Identification of a Pentapyrrolic Expanded Porphyrin in the Rothemund Synthesis[J].Chem.Eur.J.1995,1(1):68-73.
[12]游效曾,孟庆金,韩万书.配位化学进展[M].北京:高等教育出版社,2000.
[13]Robinson L R,Hambright P.Mercury(II)reactions with water-soluble porphyrins[J].Inorganic Chemistry,1992,31(4):652-656.
[14]Wu J,Liu C,Jiang Y,et al.Synthesis of chiral epichlorohydrin by chloroperoxidase-catalyzed epoxidation of 3-chloropropene in the presence of an ionic liquid as co-solvent[J].Catalysis Communications,11(8):727-731.
[15]Gao B,Boeglin W E,Zheng Y,et al.Evidence for an ionic intermediate in the transformation of fatty acid hydroperoxide by a catalase-related allene oxide synthase from the cyanobacterium Acaryochloris marina[J].Journal of Biological Chemistry,2009,284(33):22087.
[16]Zu Y,Hu N.Electrochemical detection of DNA damage induced by in situ generated styrene oxide through enzyme reactions[J].Electrochemistry Communications,2009,11(10):2068-2070.
[17]Wasielewski M R.Photoinduced Electron Transfer in Supramolecular Systems for Artificial Photosynthesis[J].Chem.Rev.1992,92(3):435-461.
[18]Baskaran D,Mays JW,Zhang X P,et al.Carbon nanotubes with covalently linked porphyrin antennae:photoinduced electron transfer[J].J.Am.Chem.Soc.,2005,127(19):6916-6917.
[19]Derege P J,Williams S A,Therien M J.Direct evaluation of electronic coupling mediated by hydrogen bonds:implications for biological electron transfer[J].Science,1995,269(5229):1409.
[20]Ming S,Zhu M H,Du Y K,Yang P,et al.The synthesis,light-harvesting,and photocatalysis of naphthylporphyrin functionalized platinum nanocomposites[J].Dyes and Pigments,2010,86:81-86.
[21]Milic T,Garno J C,Batteas J D,et al.Self-organization of self-assembled tetrameric porphyrin arrays on surfaces[J].Langmuir,2004,20(10):3974-3983.
[22]Leung W H,Che C M.High-valent ruthenium (IV)and-(VI)oxo complexes of octaethylporphyrin.Synthesis,spectroscopy,and reactivities[J].J.Am.Chem.Soc.,1989,111(24):8812-8818.
[23]Watanabe Y,Fujii H.Characterization of high-valent oxo-metalloporphyrins[J].Metal-Oxo and Metal-Peroxo Species in Catalytic Oxidations,2000:61-89.
[24]Nam W,Goh Y M,Lee Y J,et al.Biomimetic alkane hydroxylations by an iron(III)porphyrin complex with H2O2and by a high-valent iron(IV)oxo porphyrin cation radical complex[J].Inorg.Chem.,1999,38(13):3238-3240.
[25]Atkinson J K,Hollenberg P F,Ingold K U,et al.Cytochrome P450-catalyzed hydroxylation of hydrocarbons:kinetic deuterium isotope effects for the hydroxylation of an ultrafast radical clock[J].Biochemistry,1994,33(35):10630-10637.
[26]Yang Y,Chen H,Jeong Y J,et al.Nam Enhanced Reactivities of Iron(IV)-Oxo Porphyrin p-Cation Radicals in Oxygenation Reactions by Electron-Donating Axial Ligands[J].Chemistry-A European Journal,2009,15(39):1039-1046.
[27]Xiang Q,Li Y C,Kang S Z,et al.Preparation and enhanced visible light-driven catalytic activity of ZnO microrods sensitized by porphyrin heteroaggregate[J].Applied Surface Science,2010,256(2):6705-6709.
[28]蔡金华,黄锦汪,叶元坚,等.卟啉敏化二氧化钛复合微球的制备及其光催化性能[J].催化学报,2009,30(5):440-446.
[29]Huang Y,Ma W,Li J,et al.A Novelβ-CD-Hemin Complex Photocatalyst for Efficient Degradation of Organic Pollutants at Neutral pHs under Visible Irradiation[J].J.Phys.Chem.B,2003,107(35):9409-9414.
[30]Maldotti A,Andreotti L,Molinari A,et al.Photocatalytic properties of iron porphyrins revisited in aqueous micellar environment:oxygenation of alkenes and reductive degradation of carbon tetrachloride[J].Green Chemistry,2001,3(1):42-46.
[31]Gokakakar S D,Salker A V.Solar assisted photocatalytic degradation of methyl orange over synthesized copper,silver and tin metalloporphyrins[J].Indian Journal of Chemical Technology.2009,16:492-498.
[32]Harmon H J.Photocatalytic demethylation of 2,4,6-trinit-rotoluene (TNT)by porphyrins[J].Chemosphere,2006,63(7):1094-1097.
[33]Cai J H,Huang J W,Zhao P,Ye Y J,et al.Silica microspheres functionalized with porphyrin as a reusable and efficient catalyst for the photooxidation of 1,5-di-hydroxynaphthalene in aerated aqueous solution[J].Journal of Photochemistry and Photobiology A:Chemistry,2009,207:236-243.
[34]Wooyul K,Jihee P,Hwa J J,et al.Visible Light Photocatalysts Based on Homogeneous and Heterogenized Tin Porphyrins[J].J.Phys.Chem.C,2008,112:491-499.
[35]Monteiro C,Pereira M M,Aaenha M E,et al.A comparative study of water soluble 5,10,15,20-tetrakis(2,6-dichloro-3-sulfophenyl)porphyrin and its metal complexes as efficient sensitizers for photodegradation of phenols[J].Photochemical & Photobiological Sciences,2005,4(8):617-624.
[36]Chang C J,Loh Z H,Shi C,et al.Targeted proton delivery in the catalyzed reduction of oxygen to water by bimetallic pacman porphyrins[J].J.Am .Chem.Soc.,2004,126(32):10013-10020.
[37]Konstaninos C.Christoforidis M L,Elena R.Et al.Mechanism of catalytic decomposition of pentachlorophenol by a highly recyclable heterogeneous SiO2-[Feporphyrin]catalyst[J].Journal of Catalysis 2010,270:153-162.
[38]Mahdi H,Saida S,Nasser S.Selective photocatalytic epoxidation of cyclooctene by molecular oxygen in the presence of porphyrin sensitizers[J].Reac.Kinet.Mech.Cat.,2010,99:243-250.
[39]Giuseppe M,Elisa G,Leonardo P,et al.TRMC,XPS,and EPR Characterizations of Polycrystalline TiO2Porphyrin Impregnated Powders and Their Catalytic Activity for 4-Nitrophenol Photodegradation in Aqueous Suspension[J].J.Phys.Chem.,2005,B(109):12347-12352.
[40]Knoer G,Vogler A.Photochemistry and photophysics of antimony(III)hyper porphyrins:activation of dioxygen induced by a reactive sp excited state[J].Inorg.Chem.,1994,33(2):314-318.
[41]Walid M.HikalI H.James H.Photocatalytic self-assembled solid porphyrin microcrystals from water-soluble porphyrins:Synthesis,characterization and application[J].Polyhedron,2009,28:113-120.
[42]Borovkov V V,Lintuluoto J M,Sugiura M,et al.Remarkable stability and enhanced optical activity of a chiral supramolecular bis-porphyrin tweezer in both solution and solid state[J].J.Am.Chem.Soc.,2002,124(38):11282-11283.
[43]Liu C J,Yu W Y,Li S G,et al.Ruthenium meso-tetrakis(2,6-dichlorophenyl)porphyrin complex immobilized in mesoporous MCM-41as a heterogeneous catalyst for selective alkene epoxidations[J].J.Org.Chem.,1998,63(21):7364-7369.
[44]Huang H,Gu X,Zhou J,et al.Photocatalytic degradation of Rhodamine B on TiO2nanoparticles modified with porphyrin and iron-porphyrin[J].Catalysis Communications,2009,11(1):58-61.
[45]Chang Y,Hsieh Y H,Cheng T C,et al.Photocatalytic degradation of 2,4-dichlorophenol wastewater using porphyrin/TiO2F complexes activated by visible light[J].Thin Solid Films,2009,517(14):3888-3891.
[46]Bown S G,Tralau C J,Smith P D,et al.Photodynamic therapy with porphyrin and phthalocyanine sensitisation:quantitative studies in normal rat liver[J].British journal of cancer,1986,54(1):43.
猜你喜欢
文萃报·周五版(2022年19期)2022-05-17 23:01:36
艺术品鉴(2020年1期)2020-01-19 06:00:54
中成药(2018年12期)2018-12-29 12:26:08
农家科技中旬版(2018年5期)2018-07-14 01:57:58
分析化学(2017年12期)2017-12-25 12:50:09
中南大学学报(自然科学版)(2016年2期)2017-01-19 07:36:55
中国照明(2016年5期)2016-06-15 20:30:13
合成化学(2015年1期)2016-01-17 08:55:47
电源技术(2015年2期)2015-08-22 11:27:52
天然产物研究与开发(2014年7期)2014-04-27 14:16:08
