
耐氧化芳香聚酰胺纳滤膜的研究进展
2011-10-18 08:56:46张兆利郭竹洁
化工进展 2011年10期
张兆利,王 枢,郭竹洁,王 娇
(西南交通大学生命科学与工程学院,四川 成都 610031)
进展与述评
耐氧化芳香聚酰胺纳滤膜的研究进展
张兆利,王 枢,郭竹洁,王 娇
(西南交通大学生命科学与工程学院,四川 成都 610031)
芳香聚酰胺纳滤膜不耐氧化,易被活性氯氧化降解,导致膜性能急剧下降,缩短膜的使用寿命,目前已成为制约芳香聚酰胺纳滤膜应用和发展的关键问题之一。本文综述了芳香聚酰胺纳滤膜的材料和结构,重点概述了芳香聚酰胺氯化和膜性能下降的机制,并进一步介绍了近年来耐氧化芳香聚酰胺纳滤膜的研究进展。
芳香聚酰胺;纳滤膜;活性氯;氧化降解;耐氧化
纳滤膜具有高效率、低能耗、高选择性等分离特性,被广泛应用于食品、医药和环境等行业中[1-3]。芳香聚酰胺纳滤膜是目前市场上最常见的复合纳滤膜,具有脱盐率高、通量大、化学稳定性好、适用范围宽、操作压力要求低等优点,主要应用在饮用水净化、海水淡化、抗生素提取与分离、环境废水处理及催化剂分离和回收等方面。但其不耐氧化,易被活性氯氯化降解,目前已成为制约芳香聚酰胺纳滤膜应用和发展的关键问题之一[4]。
膜污染是限制纳滤膜分离技术大规模生产与应用的主要因素,主要由无机物污染、有机物污染和微生物污染构成。有机物和微生物的污染经常发生在纳滤膜分离过程,它们吸附在膜的表面,形成污染层,导致膜的性能(通量和截留率)的下降[5-6]。采用氧化剂清洗是清除有机物和微生物污染、恢复膜渗透能力的主要工艺之一。活性氯是膜过程中最常用的氧化剂或灭菌剂之一,目前芳香聚酰胺纳滤膜的抗氧化性较差,易受活性氯降解而导致膜性能的急剧下降[7]。因此,研究芳香聚酰胺氯化的机制,探索氯化导致聚酰胺纳滤膜分离性能下降的机制,并在此基础上制备高耐氯的复合纳滤膜,以期为耐氯复合纳滤膜的商品化提供理论依据。
1 芳香聚酰胺纳滤膜的应用
芳香聚酰胺纳滤膜是目前商品复合纳滤膜的主流,主要采用界面聚合法制备,通常是在超滤支撑膜上,多元胺和多元酰氯反应并形成致密的聚酰胺分离层,而后在一定温度下后处理得到成品纳滤复合膜[4]。超滤支撑膜材料主要有聚砜、聚醚砜、磺化聚砜、混合纤维素酯、聚苯醚、磺化聚醚砜、聚丙烯腈、聚氯乙烯、苯乙烯-丙烯腈、聚酰亚胺、聚对苯二甲酸丁二醇酯等[8]。
1.1 分离层的功能材料
界面聚合法能在短时间内快速发生反应形成完整而致密的复合膜[8]。因此,分离层的功能单体多选用多元胺和多元酰氯(或异氰酸酯)。为了使制备得到的聚酰胺复合纳滤膜具有一定交联度和机械性,两种单体中应至少有一个单体的官能度大于2,并且至少有一个为芳香族化合物[9]。
1.2 水相多元胺材料
制备芳香聚酰胺复合纳滤膜的多元胺类分为芳香族和脂肪族两大类。常用的芳香族多元胺有邻苯二胺(OPD)、间苯二胺(MPD)、对苯二胺(PPD)、N,N-二甲基间苯二胺(DMMPD)、4-甲基间苯二胺(MMPD)、间苯二胺-5-磺酸(SMPD)、均苯三胺(BTA)和聚间胺基苯乙烯(PmAS)等。常用的脂肪族多元胺有哌嗪(PIP)、聚乙烯醇(PVA)、1,4-环己二胺(HDA)、聚乙烯胺(PEI)壳聚糖(CS)和多胺基聚氧乙烯(MAPEG)等[9]。部分多元胺的化学结构式如图1所示。
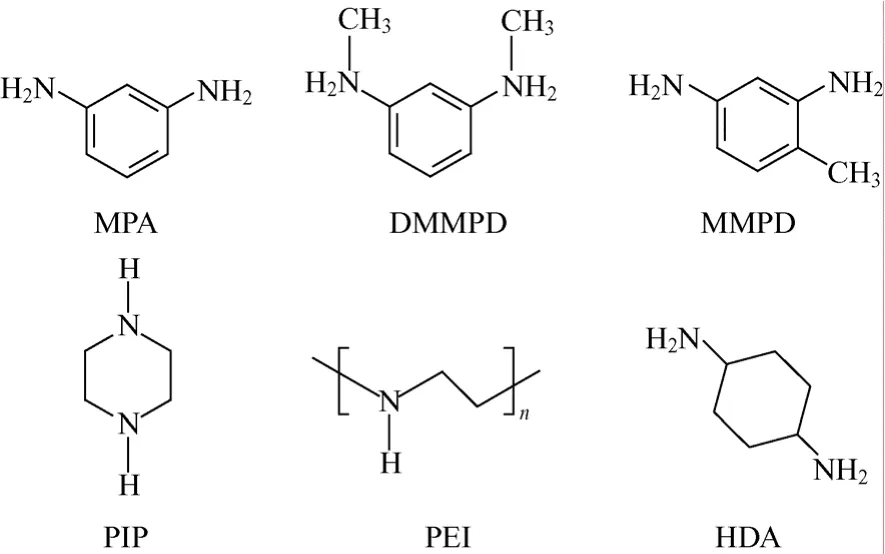
图1 多元胺的化学结构式
1.3 有机相多元酰氯(或异氰酸酯)材料
制备复合纳滤膜的有机相单体材料主要为酰氯、异氰酸酯等。常用的有机相单体有邻苯二甲酰氯(OPC)、间苯二甲酰氯(IPC)、对苯二甲酰氯(TPC)、均苯三甲酰氯(TMC)、甲基间苯二异氰酸酯(TDI)和 1,3,5-环己烷三甲酰氯(HT)、联苯三酰氯(BTRC)、联苯四酰氯(BTEC)、5-氧甲酰氯-异酞酰氯(CFIC)和5-异氰酸酯-异酞酰氯(ICIC)等[8-13]。部分多元酰氯的化学结构式如图2所示。

图2 多元酰氯的化学结构式
上述水相功能材料和有机相功能材料通过界面聚合法制备得到各种性能优异的复合纳滤膜[4,13],具有代表性的商品复合纳滤膜有:①FILMTEC公司的FT-30复合膜,采用MPD与TMC界面聚合制得,具有较高的水通量和溶质截留率;②Film Tec公司的NF-40复合膜,采用PIP与IPC/TMC界面聚合制得,具有较高的耐氯性能;③HYDECANME公司的PA-300复合膜,采用MAPEO与IPC/TDI反应制得,提高耐氯性的同时,水通量和溶质截留率均维持较高水平。
2 芳香聚酰胺膜的氯化机制
芳香聚酰胺膜不耐氧化,易被氯化降解,已成为目前制约其应用和发展的关键问题之一。因此,研究氯化处理对芳香聚酰胺膜的影响非常重要。目前的研究表明,芳香聚酰胺膜的氯化主要分为两种情况:酰胺的氯化和芳香环的氯化[14]。
2.1 酰胺的氯化
芳香聚酰胺膜氯化的第一种情况是酰胺的氯化,酰胺上的N—H键易受活性氯攻击而生成N—Cl键,是由于羰基的吸电子效应造成的。反应包括两步,即带正电的氯原子首先附着到酰胺单元上的带有孤对电子的O原子上,然后迅速发生重排而生成N—Cl键[15-16]。其反应机制如图3所示。
2.2 芳香环的氯化

图3 酰胺的氯化机理
芳香聚酰胺膜氯化的另一种情况是芳香环的氯化,目前通过对芳香聚酰胺纳滤膜的氯化研究发现,芳香环的氯化主要有两种可能的途径:①芳香环的直接氯化[17];②Orton重排发生芳香环的氯化[18]。
2.2.1 芳香环的直接氯化
芳香环氯化的一种可能的途径是芳香环直接氯化,由于芳香环属于富含电子基团,电子云密度较高,对氯非常敏感。而 Cl2为活性氯的主要成分时,芳香环和 Cl2会发生亲电取代反应,使芳香环直接氯化[17]。其反应机制如图4所示。
2.2.2 Orton重排发生芳香环氯化
芳香环氯化的另一种可能的途径是 Orton重排,芳香环的Orton重排是在酰胺氯化的基础上进行的,首先,酰胺上N—H键发生氯化生成N—Cl键,形成反应中间体,反应条件高,难度大,生成的化合物不稳定。在酸存在条件下N—Cl迅速脱氯变成N—H和Cl2,Cl2又与芳香环发生亲电取代,最终导致芳香环氯化。在 Orton重排过程中,N—Cl键是一个必要的中间体。在碱性条件下,N—Cl和N—H的转化是可逆的[18]。芳香酰胺的Orton重排机理如图5所示。

图4 芳香环的直接氯化机理

图5 芳香酰胺的Orton重排机理
Glater等[19]认为芳香聚酰胺是通过亲电取代而发生芳香环的直接氯化。他们采用苯甲醯胺苯模拟DuPont公司的B-9分离层材料,氯化处理,得到以下的结果:①随着氯化的增加,单一取代的红外吸收峰740 cm-1完全消失,并出现了对位取代红外吸收峰810 cm-1;②核磁共振氢谱分析(1H NMR)显示酰胺上N—H键未被氯化。由于胺基是芳香环亲电取代的活化基团,而羰基属于钝化基团,因此氯化主要发生在与胺基相连的芳香环上。
然而,Kawaguchi等[20]通过氯化实验却得出芳香聚酰胺通过 Orton重排而实现芳香环氯化的结论。他们考察了仲酰胺和季酰胺的氯化行为,研究在有和没有N—H存在时的氯化情况,发现季酰胺与氯没有反应,而仲酰胺发生了可逆的N—H的氯化以及不可逆的芳香环的氯化反应。结果表明,酰胺N—H基团在Orton重排过程中是重要的功能基团。首先,酰胺 N—H氯化后生成 N—Cl,N—Cl键不稳定,易脱氯而发生Orton重排使芳香环发生氯取代。并且通过1H NMR分析证实了芳香环氯取代的位置是在与胺基相连的芳香环的对位上。Lowell等[21]通过比较各种酰胺模拟化合物与NaClO的作用,也得出类似的结果。
Soice等[22]通过悬滴机械分析法(PDMA)研究MPD-TMC聚酰胺复合膜氯化降解机制,得出通过Orton重排实现芳香聚酰胺的氯化。酰胺 N—H取代为N—Cl,生成中间体,反应条件高,难度较大,中间体不稳定,在碱性条件下,可以发生可逆的反应生成N—H。N—Cl中间体迅速发生Orton重排,反应很容易进行,生成芳香环氯取代化合物。Shintani等[23-24]研究17种多元胺和多元酰氯生成的聚酰胺化合物的氯化实验,也得到相同的结果。
研究表明,聚酰胺的氯化降解主要与活性氯的主要成分及浓度密切相关,而活性氯的主要成分及浓度是由活性氯溶液的pH值和浓度密切相关[25-27]。当pH≤1或溶液中Cl2浓度较高时,Cl2为活性氯的主要成分,芳香聚酰胺的氯化机理可能是通过亲电取代而发生芳香环直接氯化。但在实际生产过程中,纳滤膜使用的pH值范围常为2~11,通常原料液中Cl2浓度很低,因此通过亲电取代发生芳香环直接氯化不是芳香聚酰胺膜氯化的主要原因。当pH值为2~8时,HClO为活性氯的主要成分,芳香聚酰胺可能是通过Orton重排而发生不可逆的芳香环的氯化,这种芳香聚酰胺的氯化机理得到当前的研究学者广泛认可[28-30]。当pH≥10时,ClO-为活性氯的主要成分,由于ClO-的反应活性较低,只能与芳香聚酰胺的末端胺基发生作用,而难以使酰胺键和芳香环发生氯化。
3 芳香聚酰胺膜氯化导致性能下降机制
芳香聚酰胺膜氯化后会导致膜的性能下降。一般认为氯化导致芳香聚酰胺膜性能下降主要有两个原因:①聚酰胺复合物的变形导致活性分离层截留性能的下降,通量增加;②酰胺键断裂而发生部分或完全降解,最终导致膜性能的降低。
聚酰胺复聚合物变形首先由Glater和Zachariah于1985年提出的,解释DuPont公司B-9膜对氯敏感的原因[19]。他们使用红外光谱对氯溶液浸泡过的膜进行分析。结果显示,红外吸收峰的波数发生明显的迁移,即从3280 cm-1变到3420 cm-1,这可能是氢键变化引起的,分子间的氢键(NH—OC)变成了分子内的氢键(NCl—H)。氢键的变化最终导致了聚合物链的变形[25]。
此外,研究氯化后芳香聚酰胺膜材料的物理性质发现,熔点、黏度、机械强度等随着氯化程度的增大而导致不同程度的下降[31-33]。这些现象均可以通过芳香聚酰胺氯化后发生降解而得到较好解释。Avlonitis等[14]则认为在碱性氯溶液中芳香聚酰胺发生的是Hoffmann降解,而在酸性氯溶液中芳香聚酰胺发生氧化降解。Koo等[34]使用电子能谱化学分析(ESCA)研究氯化破坏的FT-30膜,发现膜面的羧基含量增大,并提出了酰胺键氧化断裂的机理。
Soice等[22]还提出了另一种基于pH值变化导致聚合物链发生水解而断裂的机理,即芳香聚酰胺在通常pH值下发生氯化产生大量N—Cl键,分离层与支撑层之间的氢键遭到破坏,从而使分离层与支撑层分离;在pH>11的环境中,N—Cl键发生水解而导致聚合物链的断裂。
Soice等[27]还采用气相色谱-质谱仪(GC/MS)分析 12种模拟酰胺化合物经高浓度氯化处理后的产物,以探索酰胺键是否断裂。结果表明,只在极苛刻的氯化条件下(经15×104mg/L活性氯处理)发生了酰胺键的轻微断裂。在实际应用中,很少出现这种极苛刻的氯化条件,因此酰胺键的断裂不是引起纳滤膜性能下降的主要原因。他们认为芳香聚酰胺的氯化使得膜表面由亲水向疏水化转变,水从聚合物中挤出,膜发生消溶胀。随着氯化程度的增大,这种消溶胀作用变得不可逆,最终导致膜性能的下降。
综上所述,通过研究模拟化合物、线性结构的芳香聚酰胺及弱交联的芳香聚酰胺与活性氯相互作用[14,19,35],得出普遍接受的芳香聚酰胺氯化及导致膜性能下降的机理如下:芳香聚酰胺发生酰胺键上的N—H键氯化,并通过Orton重排而发生芳香环氯化。芳香聚酰胺复合纳滤膜的氯化可能使膜表面物理化学性质发生变化,聚合物链间氢键遭到破坏,导致链的灵活性增大,分子间氢键变成分子内的氢键,聚合物链发生变形;芳香聚酰胺的氯化会使膜表面的亲水性能下降,氢键被破坏后会使聚合物膜变得疏松,导致氯化后芳香聚酰胺膜性能的下降。在通常的pH值环境中,活性氯作用于分离层与支撑层之间,分离层与支撑层之间的氢键遭到破坏,从而使分离层与支撑层分离,导致膜性能急剧下降。另外,在极苛刻的氯化条件下或者活性氯溶液pH>11情况下,氯化后的芳香聚酰胺会发生酰胺键断裂而发生部分或完全降解,从而导致芳香聚酰胺膜性能的下降。
4 耐氯芳香聚酰胺纳滤膜的制备
通过研究和分析芳香聚酰胺膜氯化降解的机理,可以看出酰胺键的活性降低时,聚酰胺抗氯性会增强。因此,耐氯复合纳滤膜的制备主要是降低酰胺键的活性,抑制N—H氯化和Oroton重排。Soice等[22]提出了提高聚酰胺抗氯性的方法:①在酰胺的N上引入—CH3取代基以防止 N—H氯化;②在芳香氨基的所有邻位上均引入—CH3,从而增大位阻;③在苯环上引入吸电子基团,降低电子密度,阻止邻位取代,从而防止酰胺的氯化和Oroton重排。Kongaya等[36]则认为使用以下几种胺类可以有效提高抗氯性:①具有仲胺基的脂肪二胺;②在两端氨基之间甲基链短的脂肪或环脂肪二胺;③在氨基的邻位上有一个—Cl或—CH3基团的芳香胺。
目前,制备耐氯复合纳滤膜主要集中在以下 6个方面。
4.1 消除酰胺上的N—H键
制备的复合纳滤膜由于消除酰胺上的 N—H键,不含易受活性氯攻击而具有较高的耐氯性能。Shintani等[23-24]使用N,N-二甲基间苯二胺(N,NDMMPD)、N-苯基乙二胺(N-PED)等仲胺与TMC或TMC和IPC的混合物反应制备的复合纳滤膜的耐氯性能均高于商品交联芳香聚酰胺复合膜。采用N,N-DMMPD与TMC和IPC的混合物反应制备得到的纳滤膜,在40 ℃、200 mg/L的NaClO溶液下浸泡 96 h后,NaCl和Na2SO4的截留率为 95%和99.5%,能在20mg/L的NaOCl溶液中稳定运行超过70天。Kongaya等[36]制备含哌嗪结构的聚酰胺复合纳滤膜,在活性氯浓度为50 mg/L、pH=5的条件下浸泡100 h后,NaCl的截留率最高可达99.3%。Gaeta等[37]制得的聚-2,5-二甲基哌嗪噻吩酰胺膜,在活性氯浓度为3 mg/L、pH=6~7条件下至少稳定运行3年。可见,使用哌嗪、仲胺等制备复合纳滤膜,消除了酰胺上易受活性氯攻击的N—H键,改善了膜的耐氯性能,但所制备的纳滤膜的截留率一般低于交联芳香聚酰胺复合纳滤膜。
4.2 增大空间位阻
在芳香聚酰胺的芳香环的邻位或对位引入功能基团,如—CH3、—Cl等,增大空间位阻,减少活性氯对芳香聚酰胺的降解作用。Shintani等[23]研究表明,邻苯二胺制得的聚酰胺膜耐氯性优于邻苯二胺、间苯二胺和对苯二胺。Kongaya等[36]研究表明,在芳香环上氨基的邻位引入—CH3,可以提高聚酰胺的耐氯性,在50 mg/L的NaOCl溶液中稳定工作100 h以上。Kim等[38]通过烷基化反应增大了芳香聚酰胺的空间位阻,提高了聚酰胺膜的耐氯性能。
4.3 引入保护基团
引入的保护基团通常为吸电子基团,如—NO2、—COOH、—SO3H等,能降低电子密度,降低芳香环的活性,阻止邻位取代,从而阻止N氯化和Orton重排的发生[39]。Gao Congjie 等[40]用 25%的 3,3′-二氨基二苯砜和75%的4,4′-二氨基二苯砜与对苯二甲酰氯的缩聚物制得了一种耐游离氯的聚砜酰胺纳滤膜,在50 mg/L的NaOCl溶液中稳定工作超过150 h,NaCl和Na2SO4的截留率基本不变。Iborra等[41]用4,4′-二氨基二苯砜与对苯二甲酰氯和间苯二甲酰氯的聚合物的缩聚物分别制得了耐游离氯的聚砜酰胺膜,在50 mg/L的NaOCl 溶液浸泡24 h后,NaCl的截留率从 78%降到 63%~65%,水通量从 73 L/(m2·h)降到 32~38 L/(m2·h)。然而,引入的吸电子基团在提高芳香聚酰胺膜的耐氯性能同时,也降低了膜的截留性能。
4.4 脂环或脂肪胺代替芳香胺
脂环、脂肪类基团属于供电子基团,增强了酰胺键的电子云密度,从而改善了聚酰胺的耐氯性能。另外,脂环或脂肪二胺单体制备得到的聚酰胺只发生可逆的酰胺键的氯化,而不发生不可逆的芳香环的氯化。Shintani等[23]研究发现胺类制备的聚酰胺的耐氯性顺序为:芳香族二胺<脂环族二胺<脂肪族二胺。Buch等[42]使用1,3-环己二甲胺与TMC通过界面聚合法制备得到了具有较高耐氯性能的复合膜,在5 mg/L的NaClO溶液中浸泡48 h后,NaCl和Na2SO4的截留率为 97%和95.5%。Liao等[43]用1,3-二氨基-2-羟基丙烷和环氧氯丙烷的聚合物与间苯二甲酰氯交联制得的复合膜在 100 mg/L的NaClO溶液中浸泡 24 h后,膜的脱盐氯维持在90%以上,但通量有所提高。Chau等[35]用过量3-硝基苯胺和TMC形成预聚物后再与TMC交联制得复合膜,在10 mg/L NaClO溶液中浸泡60 h后,NaCl和 Na2SO4的脱盐率高达 99.6%和 99%,但制备工艺较为复杂。研究表明,脂环或脂肪胺代替芳香胺制备聚酰胺膜提高耐氯性,但引入脂环或脂肪胺后,会降低膜的机械强度,不宜在压力较高的条件下使用。
4.5 表面涂层
Kang等[30]将聚N,N-甲基丙烯酸二甲氨基乙酯(PDMAEMA)涂敷于聚酰胺膜表面,形成一层保护层,PDMAEMA先与活性氯反应,减少活性氯对膜面活性位的攻击。在5 mg/L的NaClO溶液中浸泡 1个月后,膜截留率下降约 3.5%。然而,涂敷PDMAEMA层后,膜通量有所下降,并且易被稀酸破坏。此外,商品纳滤膜(如LFC1、LFC3、BW30和SW30HR)表面涂覆PVA后,不仅改善了膜抗污染性能,也使膜的耐氯性能有所提高[44-45]。然而,表面涂层的方法却往往导致纳滤膜的通量下降。
4.6 开发新型材料
Jayarani等[46]用聚酯制得的膜的耐氯性优于芳香聚酰胺膜,在10mg/L的NaClO溶液中浸泡240 h后,NaCl和Na2SO4的截留率为96%和99.2%,膜通量基本不变。Kim等[38]用多元酚的水溶液与TMC反应制备得到的膜具有较高耐氯性能和选择透过性能。浸泡在200 mg/L的NaClO溶液中96 h后,Na2SO4的截留率为 98.9%,膜通量基本不变。Konagaya等[47]用3,3-二氨基二苯砜和对苯二甲酰氯及 3,5-二氨基苯甲酸的共聚物制得到的膜具有优良的耐氯性能和选择透过性能。在pH=5的条件下,用220 mg/L的NaClO溶液处理24 h后,海水的脱盐率为 98.3%,通量为 159 L/(m2·d)。Park 等[48]合成磺化双酚型聚芳香醚砜(BPS)膜,易于控制磺化度,具有很好的耐氯性、抗水解性和机械稳定性。由BPS制得到的膜在pH值为4~10范围内具有很好的耐氯性能。在活性氯浓度 500 mg/L、pH=9.5下浸泡 20 h的条件下,商品交联芳香聚酰胺膜的Na2SO4截留率下降了20%,而BPS膜(40%磺化度)的盐截留率没有明显变化,水通量较高。采用新型材料制备复合膜,是耐氯芳香聚酰胺纳滤膜研究制备的一个新的方向,能提高纳滤膜的耐氯能力,但性能很好的工业化商品并不多。
综上所述,目前制备耐氯复合纳滤膜主要集中在以上6个方面,并且在实验研究阶段都取得了一定的成绩,都能够一定程度上改善复合纳滤膜的耐氯性能。然而,以上6种方法并不能较大幅度地提高复合膜的耐氯时间和强度,也会影响膜的选择透过性能。消除酰胺上的N—H键,能改善复合纳滤膜的耐氯性,但同时也降低了膜的溶质截留率,导致膜的分离性能下降。引入—NO2、—COOH、—SO3H等吸电子基团作为保护基团后,也会出现类似的情况。引入—CH3、—Cl等功能基团,增大空间位阻,减少活性氯对芳香聚酰胺的降解作用,能够提高复合纳滤膜的耐氯性,然而,增大空间位阻的同时,也降低了胺类的反应活性,导致界面聚合的难度增加,最终影响复合膜的制备。脂环、脂肪胺等供电子基团代替芳香胺,增强了酰胺键的电子云密度,从而改善了聚酰胺的耐氯性能,但引入脂环或脂肪胺后,会降低膜的机械强度,不能在压力较高的条件下使用。表面涂层是一种新兴的复合膜改性方法,膜的耐氯性能有所提高,然而此方法往往导致纳滤膜的通量下降。采用新型材料制备复合膜,是耐氯芳香聚酰胺纳滤膜研究制备的一个新的方向,能提高纳滤膜的耐氯能力,但性能很好的工业化商品并不多。
5 结 语
目前芳香聚酰胺纳滤膜已经广泛应用到食品、医药、化工和环境等领域,并且取得了较好的经济效益和社会效益。但是对耐氯芳香聚酰胺纳滤膜的研究尚处于起步阶段。当前研究主要集中在膜材料的改性及新型耐氯膜材料的合成方面,虽然取得了一定的成绩,但性能很好的工业化商品并不多。
因此,需要进一步研究芳香聚酰胺膜的氯化降解机理,在此基础上开发新型的耐氯膜材料及纳滤膜改性制备工艺,并围绕简化工艺、降低成本、提高耐氯时间和强度,通过多学科交叉研究,从而更有效地提高纳滤的耐氯性,并兼顾膜的机械性和选择透过能,制备高耐氯、高效且廉价的复合纳滤膜。
[1]Jie Xingming,Cao Yiming,Li Hongjian,et al.Preparation and separation performance of cellulose membrane from direct dissolution[J].Journal of Chemical Industry and Engineering(China),2006,57(8):1756-1762.
[2]Su Xiaojian,Huang Lijie.Study on MF-RO combined membranes process in concentrating grosvenori infusion[J].Membrane Science and Technology,2009,29(1):66-68.
[3]Feng Xiao,Ren Nanqi,Chen Zhaobo.Performance of ultrafiltration membrane technology in treatment of soy whey wastewater[J].Journal of Chemical Industry and Engineering(China)(Ⅰ),2009,60(6):1477-1486.
[4]Robert J Peterson.Composite reverse osmosis and nanofiltration membrane[J].J. Membr Sci.,1993,83:81-150.
[5]Baker R W.Membrane technology and applications[M].Second edition.Chichester:John Wiley & Sons Ltd., 2004.
[6]时钧,袁权,高从堦.膜技术手册[M].北京:化学工业出版社,2001.
[7]Reddy A V R,Trivedi J J,Devmurari C V,et al.Fouling resistant membranes in desalination and water recovery[J].Desalination,2005,183:301-306.
[8]汪锰,王湛,李政雄.膜材料及其制备[M].北京:化学工业出版社,2003.
[9]李兆魁,周勇,朱家民,等.纳滤膜功能材料研究进展[J].水处理技术,2009,(35)12:1-5.
[10]Yu S,Liu M,Liu X,et al.Performance enhancement in interfacially synthesized thin-film composite polyamide-urethane reverse osmosis membrane for seawater desalination[J].J. Membr. Sci.,2009,342:313-320.
[11]Li L,Zhang S,Zhang X,et al.Polyamide thin film composite membranes prepared from isomeric biphenyl tetraacyl chloride andm-phenylenediamine[J].J. Membr. Sci.,2008,315:20-27.
[12]高从堦.芳香族聚酰胺纳滤复合膜的研究进展[J].第六届全国膜与膜过程报告会,天津,2008.
[13]Larson R E,Cadotte J E,Petersen R J.The FT-30 seawater reverse osmosis membrane-element testresults[J].Desalination,1981,38:473-483.
[14]Avlonitis S,Hanbury W T,Hodgkiess T.Chlorine degradation of aromatic polyamide[J].Desalination,1992,85:321-334.
[15]Jensen J S,Lam Y F,Helz G R.Role of amide nitrogen in water chlorination:Proton NMR evidence[J].Environ. Sci. Technol.,1999,33:3568-3573.
[16]Challis B C,Challis J A.Reaction of the carboxamide group//J.Zabicky(ed.),The Chemistry of Amides[M]. New York:Willey Interscience,1970:775-779.
[17]邢其毅.基础有机化学[M].第2版. 北京:高等教育出版社,2003.
[18]Orton K J P,Soper F G,Williams G.The chlorination of anilides.PartⅢ.N-chlorination andC-chlorination as simultaneous side reactions[J].J. Chem. Soc.,1998,998-1005.
[19]Glater J,Zachariah M R.A mechanistic study of halogen interaction with polyamide reverse-osmosis membranes[J].ACS Symp. Ser.,1985,281:345-358.
[20]Kawaguchi T,Tamura H.Chlorine-resistant membrane for reverse osmosis Ⅱ.Preparation of chlorine-resistant polyamide composite membranes[J].J. Appl. Polym. Sci.,1984,29:3369-3379.
[21]Lowell J R,Friesen D T,McCray S B,et al.Model compounds as fredictors of chlorine sensitivity of interfacial polymer reverse osmosis membranes[C]//Proceedings of the 1987 International congress on Membranes and Membrane Processes,Tokyo,1987.
[22]Soice N P,Alan R,Greenberg,et al.Studies of oxidative degradation in polyamide RO membrane barrier layers using pendant drop mechanical analysis[J].Journal of Membrane Science,2004,243(1-2):345-355.
[23]Shintani T,Matsuyama H,Kurata N.Development of a chlorineresistant polyamide reverse osmosis membrane[J].Desalination,2007,207:340-348.
[24]Shintani T,Matsuyama H,Kurata N.Development of a chlorine-resistant polyamide nanofiltration membrane and its field-test results[J].J. Appl. Polym. Sci.,2007,106:4174-4179.
[25]Glater J,McCutchan J W,McCray S B,et al.Effect of halogens on the performance and durability of reverse-osmosis membranes[J].ACS Symp. Ser.,1981,171-190.
[26]Glater J,McCutchan J W,McCray S B,et al.Halogen interactions with typical reverse osmosis membranes[J].Proc. Water Reuse.Symp.,1982,1399-1409.
[27]Soice N P,Maladono A C,Takigawa D Y,et al.Oxidative degradation of polyamide reverse osmosis membrane:Studies of molecular model compounds and selected membranes[J].J. Appl.Polym. Sci.,2003,90:1173-1184.
[28]Kwon Y N,Leckie J O.Hypochlorite degradation of crosslinked polyamide membranes Ⅰ.Changes in chemical/morphological properties[J].J. Membr. Sci.,2006,283:21-26.
[29]Kwon Y N,Leckie J O.Hypochlorite degradation of crosslinked polyamide membranes Ⅱ.Changes in hydrogen bonding behavior and performance[J].J. Membr. Sci.,2006,282:456-464.
[30]Kang G D,Gao C J,Chen W D,et al.Study on hypochlorite degradation of aromatic polyamide reverse osmosis membrane[J].J.Membr. Sci.,2007,300:165-171.
[31]Gabelich C J,Frankin J C,Gerringer F W,et al.Enhanced oxidation of polyamide membranes using monochloramine and ferrous iron[J].J. Membr. Sci.,2005,258:64-70.
[32]Tessaro I C,Silva J B,Wada K.Investigation of some aspects related to the degradation of polyamide membranes:Aqueous chlorine oxidation catalyzed by aluminum and sodium laurel sulfate oxidation during cleaning[J].Desalination,2005,18:275-282.
[33]Singh R.Polyamide polymer solution behavior under chlorination conditions[J].J. Membr. Sci.,1994,88:285-287.
[34]Koo J Y,Petersen R J,Cadotte J E.ESCA characterization of chlorine-damaged polyamide reverse osmosis membrane[J].Polym.Prepr.,1986,27:391-392.
[35]Chau Michael M,Light William G,Calif A,et al.Chlorine-tolerant thin-film composite membrane:US,5271843[P].1993.
[36]Kongaya S,Kuzumoto H,Watanabe O.New RO membrane materials with higher resistance to chlorine[J].Journal of Applied Polymer Science,2000,75:1357-1364.
[37]Gaeta S N,Petrocchi E,Negri E,et al.Chlorine resistance of polypiperazineamide membranes and modules[J].Desalination,1991,83:383.
[38]Kim J,Kwak S,Kim C.Composite reverse osmosis membranecomprising polymeric porous support,with an ultra thin aromatic polyester or polyester-polyamide copolymer layer:US,5593588[P].1997.
[39]Murphy A P,Murugaverl B,Riley R L.Chlorine resistant polyamide for chlorine resistant membrane,comprises reaction product of amine,and acid chloride monomer modified with electronwithdrawing groups that exhibit sufficient activity to minimize chlorination:US,5277333[P].2008.
[40]Gao Congjie.PSA asymmetric membranes[J].Desalination,1991,83(1-3):271.
[41]Iborra M I,Lora J,Alcaina M I,et al.Effect of oxidation agents on reverse osmosis membrane performance to brackish water desalination[J].Desalination,1996,108(1-3):83-89.
[42]Buch P R,Mohan D J,Reddy A V.Preparation,characterization and chloride stability of aromatic-cycloapliphatic polyamide thin film composite memberane[J].J. Membr. Sci.,2008,309:36-44.
[43]Liao Sung K,Kansas City,Chappelow Cecil B,et al.Composite reverse osmosis membrane comprising crosslinked poly(amine-epihalohydring adduct):US,4659475[P].1987.
[44]Uemura T,Kurihara M.High performance semipermeable composite membrane and process for producing the same:US,4559139[P].1983.
[45]Miller D R,Peppas N A.Diffusional effects during albumin adsorption on highly swollen poly(vinyl alcohol)hydrogels[J].Eur.Polym. J.,1988,24:611-615.
[46]Jayarani M M,Kulkami S S.Thin-film composite poly (esteramide)-based membrane[J].Desalination,2000,130:17-30.
[47]Konagaya S,Tokai M,Kuzumoto H.Reverse osmosis performance and chlorine resistance of new ternary aromatic copolyamides comprising 3,4-diaminodiphenylsulfone and a comonomer with a carboxyl group[J].J. Appl. Poly. Sci.,2001,80:505-513.
[48]Park H B,Freeman B D,Zhang Z B,et al.Water and salt transport behavior through hydrophilic-hydrophobic copolymer membranes and their relations to reverse osmosis membranes[J].PMSE Preprints,2006,95:889-891.
Development of the oxidation resistance of aromatic polyamide thin-film composite nanofiltration membranes
ZHANG Zhaoli,WANG Shu,GUO Zhujie,WANG Jiao
(School of Life Science and Engineering,Southwest Jiaotong University,Chengdu 610031,Sichuan,China)
Aromatic polyamide thin-film composite(TFC)nanofiltration(NF)membranes are very sensitive to a chlorine disinfectant. The oxidative degradation takes place under active chlorine conditions. The free chlorine in feed stream can cause degradation of crosslinked aromatic polyamides and ultimately result in the decline of the membrane separation performance,which shortens the membrane life and greatly limits the further application and development of NF membranes. Series studies of NF materials and structures in recent years are briefly summarized. This review is focused on the oxidative degradation of aromatic polyamides and the mechanism of the decline of the membrane separation performance. The prospect on the development of the oxidation resistance of aromatic polyamide thin-film composite nanofiltration membranes is further analyzed.
aromatic polyamide;nanofiltration membranes;active chlorine;oxidative degradation;oxidation resistance
TQ 028. 8
A
1000–6613(2011)10–2240–07
2011-03-28;修改稿日期2011-05-31。
国家自然科学基金(20706046)及中央高校基本科研业务费专项资金(SWJTU11CX112)项目。
张兆利(1987—),男,硕士研究生,从事膜分离研究。联系人:王枢,副教授。E-mail wone_su@163.com。
猜你喜欢
现代塑料加工应用(2021年5期)2021-02-28 08:18:26
分析化学(2019年3期)2019-03-30 10:59:24
纺织科学研究(2017年4期)2017-05-17 04:00:03
西南国防医药(2016年6期)2016-12-01 06:00:58
中国塑料(2016年1期)2016-05-17 06:13:10
中国塑料(2016年5期)2016-04-16 05:25:36
生物化工(2016年4期)2016-04-08 10:26:27
中国塑料(2015年3期)2015-11-27 03:42:15
中国塑料(2015年12期)2015-10-16 00:57:21
山东工业技术(2015年6期)2015-07-27 00:53:22
